
Abstract
Rabies vaccine preparations are quantitatively assayed for potency using the in-vivo challenge National Institute of Health (NIH), the main test that consumes a high number of animals, takes a long time, and has wide variability. The Rapid focus fluorescent inhibition (RFFIT) and the passive hemagglutination (PHA) tests, the two serologically based tests, were also used for such purpose. In this study, we aimed to evaluate and correlate the potency of the NIH, RFFIT, and PHA tests according to the World Health Organization (WHO) validity criteria, aiming to validate the use of RFFIT or PHA test as a substitute to the NIH test for determining the potency of commercially available Rabies vaccine preparations. The results showed that, the three tests can be successfully used; however, a higher correlation between RFFIT and NIH than PHA and NIH was recorded (Pearson correlation = 1). The potency of rabies vaccine preparations using NIH, RFFIT, and PHA were 3.73, 3.51, and 4.50, respectively. NIH is the main test for the determination of vaccine potency carried out by conducting 25 experiments and consuming about 5,000 mice compared to 1,200 mice used with RFFIT and 1,000 mice used with PHA test. Taken together, we concluded that (i) in some tested preparations, both RFFIT and PHA tests gave comparable results, and they can be used interchangeably; (ii) RFFIT could successfully replace NIH test, but not PHA; (iii) RFFIT and PHA tests are faster, more accurate, more economic, and more sensitive than NIH; nevertheless, PHA needs further investigations; and (iv) both RFFIT and NIH tests complement and reinforce each other as they provide a comprehensive picture of the product potency.
Introduction
Rabies is considered one of the most virulent viral infections of warm-blooded animals and still thousands of people are infected with it each year worldwide (18). It is a fatal disease unless early treatment is received before symptom appearance (17). The virus usually enters the human body through a bite from a rabid animal's saliva. Other ways include inhalation of the aerosolized rabies virus (RABV), particularly with the laboratory workers (4). About 24,000 people die due to rabies in Africa each year and the infection is mainly transmitted by dogs (18). Fortunately, it is a vaccine-preventable disease (25) and the most important way to prevent it is vaccination of both pets and people (19). Rabies vaccine is an inactivated virus vaccine prepared either in human diploid cell culture or in purified chick embryo cell culture (19).
Rabies vaccination is administrated in two cases: (i) as pre-exposure prophylaxis in persons likely to be exposed to rabies who may be at high risk due to geographical location and (ii) after exposure to animals known or suspected to be rabid. The pre-exposure immunizing course is three doses of rabies vaccine given intramuscularly on day 0, 7 days, and 21 to 28. The three-dose regimen leads to formation of protective levels of antibodies in virtually 100% of vaccinees (19). The same vaccine is used for postexposure prophylaxis or pre-exposure to rabies; therefore, high-quality control must be applied to ensure its safety, efficacy, and potency.
Potency test is an important tool for experiencing the actual relative strength of manufactured assembly lots of vaccine. Because of high variability of biological products, potency is an effective tool that assures lot-to-lot consistency of commercial vaccines (13).
The available potency assays for rabies vaccine preparations can be divided into four different categories, including (i) challenge test; (ii) toxin neutralizing test; and (iii) cell-based assays and titration assays (10). Quality control of rabies vaccine preparations should be conducted at two levels, by the manufacturer and by a national control authority.
The methods for quality control of rabies vaccines were revised by the WHO Expert Committee (24). In Egypt, Egyptian Drug Authority (EDA) is the national control authority that is responsible for testing rabies vaccine efficacy, safety, and potency. Safety and potency are the most important tests whose results must be conformed so that the product is approved for release to the public. However, the potency test is the most important test of all, as it determines the product capacity to protect against rabies disease (24).
As in whole authorities, Mouse Challenge National Institute of Health (NIH) test is the main reference test used for potency evaluation (20). This test is dependent on immunization of large number of mice, followed by challenging them with RABV. One month later, both the live and dead animals are counted followed by calculating the 50% lethal dose of the vaccine and its potency (10). In this test, determination of an immune response in an animal model would allow the rational development of an effective vaccine. However, proving this correlation is difficult.
These animals only represent the interactions of the organism with immune system (10). In addition, the main limitations of this test are as follows: consuming large number of animals, non-human endpoint for the test, time consuming, and the high variability (10,19). In addition, safety wise, risk management is easier in cell culture laboratory than in animal facility.
Even though all procedures are done in safety cabinets and animals are housed in individually ventilated cages, the virulent material injected and technicians exposed to contaminated animals increase the risk probabilities (10). Because of all these disadvantages, and because of the many approaches for replacement and/or reduction of laboratory animals in tests, there are many alternative techniques such as serological assays for potency evaluation.
Serological assays measure antibody levels produced by animals, which are representative to vaccine potency (10). So, instead of depending on number of live animals in challenge test, we depend on antibody titer. However, this test only reflects the immune response in the first phase (10).
Rapid focus fluorescent inhibition test (RFFIT) is a serological test for potency determination by measuring the levels of rabies virus neutralizing antibody titers induced after vaccination of mice against Rabies. It is a WHO standard method assay used to evaluate the immunity effect of mice after vaccination (28). Passive hemagglutination (PHA) is another serological test that was developed to overcome the disadvantages of RFFIT, being cheaper (2) and not affected by the cell type in which the virus was grown (11).
Generally, PHA's principle depends on the hemagglutinating property of RABV through combination of coupling agents such as chromium chloride (15). Mainly, the virus glycoprotein is coupled to erythrocytes from mice, or sheep, or guinea pig through this coupling agent (27). This technique is developed to overcome problems of other tests, either NIH test or neutralization assays (1). We can use any type of erythrocyte, but it is documented that goose erythrocytes were agglutinated with five strains of RABV grown on cell culture regardless of cell culture type (11). The test is able to measure antibody titer after vaccination.
The three tests used in this study were concerned with measuring the vaccination response; NIH measured the vaccination response through survival rate of mice after vaccination, and then administration of challenge virus, while RFFIT measured it through inhibition rate of lethal RABVand PHA measured the response through agglutination activity of the virus
Antibodies play a crucial rule in rabies disease prophylaxis against many infectious agents, especially RABV. Neutralization ability of antibody to inactivate infectivity of virus is the first and most important function of it. Protection against viral pathogens in vivo is sophisticated. It is normally measured in vitro. Even though it has a partial contribution in protection (14), the inability to compete infection is astonishing since both pre-exposure vaccination and—if given adequately—postexposure vaccination are highly effective at preventing rabies disease with its drastic consequences. The main immunological protection participant produced by vaccination is neutralizing antibody (26).
After infection, the main participant of protection is the presence of neutralizing antibody. There is clear proof for antigen presentation and rapid drainage from the spine and central nervous system to local lymphoid tissue, which could postpone the production of antigen to appropriate B lymphocytes (14). All the above is talking about humoral immune response against RABV. On the other hand, cellular immune response is started with plasma cells that are detected from day 7 to 14, peaking on day 10, while memory B cells appear from day 10 up to at least day 28 (16).
Humoral immunity represented in neutralizing antibody is the most important in competing RABV (22). According to the WHO, rabies virus neutralizing antibody (RVNA) titers of ≥0.5 IU/mL are considered sufficient for rabies protection. Therefore, detection and quantification of RABV antibodies are critical and of relevant importance (22). Vaccine potency is a significant factor in determining the effectiveness of RABV vaccines (8). In 1959, Russell and Burch have founded the 3Rs approach as a guiding principle for using laboratory animals ethically (mainly mice). This approach encourages reduction of animal number, its replacement, and refinement for pain alleviation.
NIH test consumes large number of animals, takes a long time, and has wide variability and only a few studies were found in literature for correlation of this test with other serological tests. Therefore, the aim of this study was to evaluate and correlate the potency of NIH, RFFIT, and PHA tests according to the WHO validity criteria, aiming to validate the use of RFFIT or PHA test as a substitute for NIH test, for determining the potency of the commercially available rabies vaccine preparations.
Materials and Methods
Experimental animals
The animals used in the study fulfilled the criteria stated in European pharmacopeia monograph 0216 under Rabies vaccine. White Swiss Albino mice weighing 10–16 g were immunized and challenged in case of NIH test and immunized only in case of RFFIT and PHA tests. Mice were quarantined for 1 week before the day of the test started, to adapt to the conditions. The whole study was approved by the Research Ethics Committee (REC) of Faculty of Pharmacy, Ain Shams University (FPREC-ASU) under approval number (No. 50).
Chemicals
Challenge virus standard was supplied frozen as 20% mouse-brain suspension in a diluent containing 2% of fetal bovine serum (FBS) in distilled water and stored in solid carbon dioxide (dry ice) at (−70°C). Sample diluent: saline, phosphate buffered saline (PBS), or sterile medium containing 2% FBS were needed for the three tests. Fluorescein goat anti-mouse IgG (H+L) cross adsorbed secondary antibody or Fluorescein Isothiocyanate (FITC), acetone (80% diluted with water), was needed for RFFIT test. Both mouse red blood cells and chromium chloride were used for PHA test. Other chemicals used in this study were of analytical reagent grade and supplied in high quality from available commercial sources.
Test samples
Rabies vaccine preparations from two different companies were used for conducting the comparative study between RFFIT and PHA tests, the three methods for potency evaluation.
RFFIT test
Potency of different tested rabies vaccine products was determined by RFFIT test as described below.
Preparation of test controls
Four controls were prepared: reference serum, cell control, virus back titration, and internal standard. The aim of preparation of these controls was to avoid errors and assure accuracy of the test.
Vaccination of test animals
Four groups, 10 mice each weighing 13–16 g, were injected intraperitoneally with 0.1 mL (1/5 of the human dose) “Verorab” the working reference vaccine (Sanofi Pasteur, Zuellig Pharma, MIMS, Malaysia). One week separated the two doses of immunization. Control mice for an adequate titration of the challenge virus were made with 10 mice for each dilution of the virus (a total of 30–40 control mice).
Preparation of sample
After 14 days of vaccination, blood was collected from each group through submandibular vein, which allows maximum allowable sample volume with minimal trauma to the animal (23), and centrifuged at 3,000 rpm/min for 30 min. It was then stored at −20°C after inactivation for 30 min at 56°C in a water bath. Serial dilution of sample using Eagle's minimal essential medium (EMEM) diluent in eight-well Lab Tek slides was carried out; the dilutions were 1:25, 125, 625, and 3125. The final volume was 0.1 mL. These samples were then mixed with 0.1 mL of challenge virus (23).
Preparation of working virus dilution and cell suspension
The same working stock virus was used in NIH test, used in this test, to avoid change in virus proteins. It was diluted with EMEM to obtain 50 fluorescent infectious activity (FFID) 50/0.1 mL. Then, 0.1 mL of working solution was added to all wells, except for cell control. The mixture of serum/virus was then incubated in CO2 incubator at 37°C for 90 min.
For cell suspension, the final concentration must be 5 × 105 cells/mL; dilution factor is calculated by dividing the initial concentration of cell suspension by the desired concentration. About 0.2 mL of cell suspension was needed in each well. One milliliter diethylamino ethyl (DEAE)- dextran was added 15 min before addition of the cell suspension of 100 mL, and then 0.2 mL of cell suspension was added to all wells. Slides were shaken carefully to mix the components and were incubated for 20–24 h in a CO2 incubator.
Titer measurement
For measuring RABV titer, tenfold dilution was made (10−1 to 10−8); 0.1 mL of each dilution was added to well of the chamber slide and incubated at 37°C for 90 min in CO2 incubator. Then 0.2 mL of cell suspension was added, containing 2.5 × 105 cells/mL, to well of the chamber slide and incubated at 37°C for 40 h in CO2 incubator (Hera cell, EDA, Cairo, Egypt). The slide was fixed, stained, and evaluated for RABV-infected cell, as mentioned under slide fixation section.
Slide staining
Chamber slides were inverted and the liquid contents were discarded after cell growth was completed. Cell monolayer was rinsed with PBS and fixed in cold acetone for 10–30 min. the slide was air dried, and anti-rabies FITC conjugate was added to cover the entire well surface (“0.16 mL” was sufficient). After incubation for 30 min at 37°C, the slide was rinsed with PBS, then water, and was dried.
Microscopic evaluation
Slides were evaluated under fluorescence microscope, 400 × and 20 microscopic fields per well were read. The virus that had not neutralized serum with RVNA-infected cells was identified by anti-rabies FITC conjugate. Pictures were taken using fluorescence microscope (LEICA DM 5500 B, El-Azhar University, Cairo, Egypt). Each well containing serum dilution was searched for fluorescing foci by observing 20 microscopic fields.
PHA test
Potency of different tested rabies vaccine products was determined by PHA test as described below.
Standard challenge virus preparation and LD50 determination
The same virus was used in NIH and RFFIT tests.
Coupling of virus and red blood cells
Mice erythrocytes were coupled with virus to form complex by chromium chloride.
PHA titration
Inactivating tested sera was added at 56 °C for 30 min, and then equal volume of mice erythrocytes was added and incubated at 37°C for 30 min. Twofold serial dilutions were made in V-shaped microtiter plates at volumes of 0.025 mL. Erythrocytes were then added to each well and plates were sealed with cellulose tape and stored at 4°C for an hour, and were examined for hemagglutination.
Reading the plate
Sharp button in the bottom of the well indicated a negative test, while a ring formation on sides of the well was a positive indication. Depending on degree of agglutination, wells were graded as “+, ++, +++, and ++++.”
NIH test
NIH test was determined for some selected rabies vaccine products using Swiss mice, weighing 14–16 g. This test relied on vaccination of mice with two doses of vaccines with a 1-week separation between the two doses, then challenging the mice with challenge RABV strain. The dead and live mice were then counted on day 28 from start of the test. The results of the test were interpreted as described in European pharmacopoeia monograph 0216.
Effect of number of dilutions used for potency determination of different vaccine samples
Three dilutions with four regimens were tested against the fourth original dilution used for potency determination as follows: Regimen 1: The first three dilutions only were used in the ED50% (effective dose 15%; a vaccine dose that gave 50% protection of the tested animals) calculations, which were further used in potency determination. Regimen 2: The last three dilutions were only used in the ED50% calculation which were further used in potency determination. Regimen 3: The first dilution and last two dilutions were used in the ED50% calculations, which were further used in potency determination. Regimen 4: The first two dilutions and the last one were used in the ED50% calculations, which were further used in potency determination.
Validity criteria were met in regimen 2, 3, and 4 with 100% of all samples and were not met in regimen 1 with 100% for all samples since the last dilution survival rate is more than 30% of total number of mice.
Results
NIH verification and potency evaluation
Determination of LD50 used in the test and challenge virus for test and control
The LD50 used in the test should be calculated using Spearman and Kärber method and it was satisfactory if it was in the range between 5 and 100 as shown in Table 1.
Determination of the Challenge Virus Strain Titer
DF: Dilution Factor, D5: 5th day of challenge, D14: the last day of test, LD50%: 50% of lethal dose of the virus.
The death on day 5 was the time when RABV becomes effective.
Relative number of dead mice D14/D5.
RABV, rabies virus.
Verification of NIH challenge test
Verification was done by the National Institute for Biological Standards and Controls (NIBSCs) reference, National Reference of China, and highly qualified Working Reference standard of Rabies vaccine (Verorab) (Sanofi Pasteur, Zuellig Pharma, MIMS, Malaysia) used as house working reference according to European Pharmacopia.
As revealed in Table 2, the results showed that the ED50% of different vaccine reference preparations was similar (2.7), while the corresponding potencies of these preparations were different (4, 3.3, 4.5 IU/dose for NIBSC, national reference of China, Working Reference standard “Verorab,” respectively). The difference in the calculated potency values of these preparations was due to the difference in labeled potency of these preparations. However, all these calculated potency values are valid (not less than 2.5 IU/dose as stated in European Pharmacopeia).
Potency of Different Vaccine Reference Preparations as Determined by National Institute of Health Challenge Test
ED50: 50% effective dose. NIBSC, National Institute for Biological Standards and Controls reference; “Verorab,” Working Reference standard of Rabies vaccine (Sanofi Pasteur, Zuellig Pharma, MIMS, Malaysia).
Vaccination was at D0, D7; and challenge was after 14 days from first vaccination.
Potency evaluation using NIH challenge test
In this test, 22 different vaccine samples manufactured by a single company were tested by NIH challenge test. The results are delineated in Table 3.
Calculated ED50% and Potency of Different Tested Vaccine Preparations as Determined by National Institute of Health Challenge Test
ED50: 50% effective dose.
Effect of number of dilutions used for potency determination of different vaccine samples
Different regimens were tested using dilutions as mentioned in 2.6.1. Acceptance criteria—according to EP—were met in all samples regarding all regimens, since in no case potency was less than 2.5 IU/mL and the results are shown in Table 4. Regimen 1 is the original one that is used in the tests where all dilutions were considered in the ED50% calculations and in the vaccine potency determination. Only 22 out of 25 samples in regimen 2 give the same results of original regimen, while the rest of other regimens give different results from the original one. However, all of them still conformed samples. In regimen 1, only 3 samples give similar results as the original one. The fold change, on average onefold change (0.7–1.3) in different regimens using different dilutions in ED50% and potency, was also calculated.
Different Regimens of Using Different Dilutions
Regimen 1: using the first three dilutions.
Regimen 2: using the last three dilutions.
Regimen 3: using the first dilution and last two.
Regimen 4: using the first two dilutions and the last one; Verorab: working reference standard of Rabies vaccine (Sanofi Pasteur, Zuellig Pharma, MIMS, Malaysia).
RFFIT verification and potency evaluation
RFFIT verification
In this experiment, the working standard human rabies immunoglobulin (CSL Behring, Zuellig, MIMS, Philippines) “Berirab” was used. Four validation parameters were chosen for this verification process; specificity that recorded 88%, sensitivity that was 80%, variant coefficient results, and repeatability are shown in Table 5 and Figure 1. The repeatability of RFFIT test is shown in Table 6. Microscopic evaluation of the RABV neutralization using RFFIT is displayed in Figure 2.
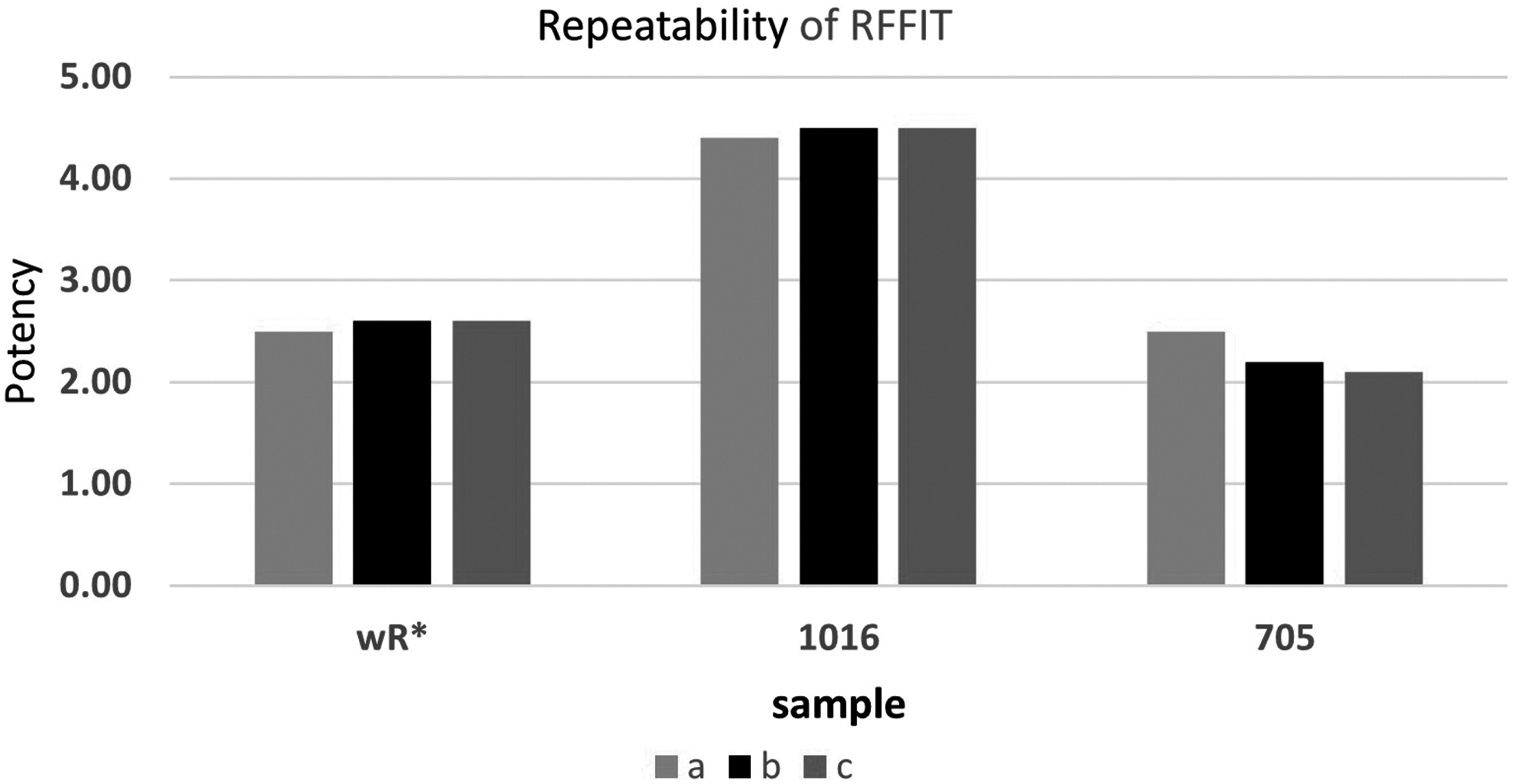
Repeatability of RFFIT. *wR, working reference. RFFIT, rapid focus fluorescent inhibition test.
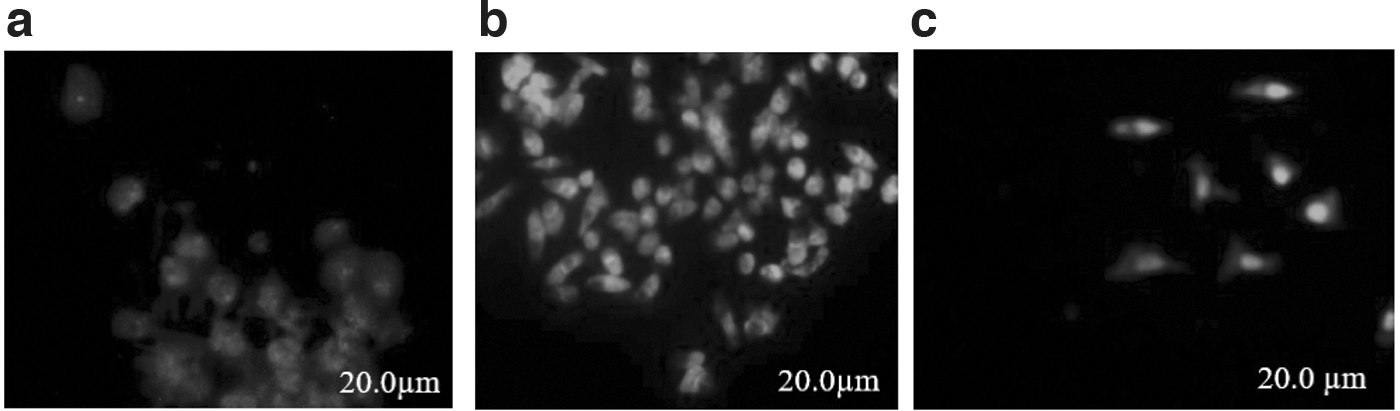
Microscopic evaluation of virus neutralization using RFFIT:
Results Rapid Focus Fluorescent Inhibition Test Verification
According to FDA, ICH guidelines.
CV, coefficient of variance.
Repeatability of Rapid Focus Fluorescent Inhibition Test
“Berirab,” working standard human rabies immunoglobulin (CSL Behring, Zuellig, MIMS, Philippines.
SD, standard deviation.
Potency evaluation using RFFIT
Rabies vaccine preparations used in RFFIT are 22 samples of rabies vaccine of two companies (Vero cell). Results of potency evaluation in the test are shown in Table 7. Comparison between survival rate of NIH test and antibody titer of RFFIT for Verorab (Reference standard of rabies vaccine) is shown in Figure 3.
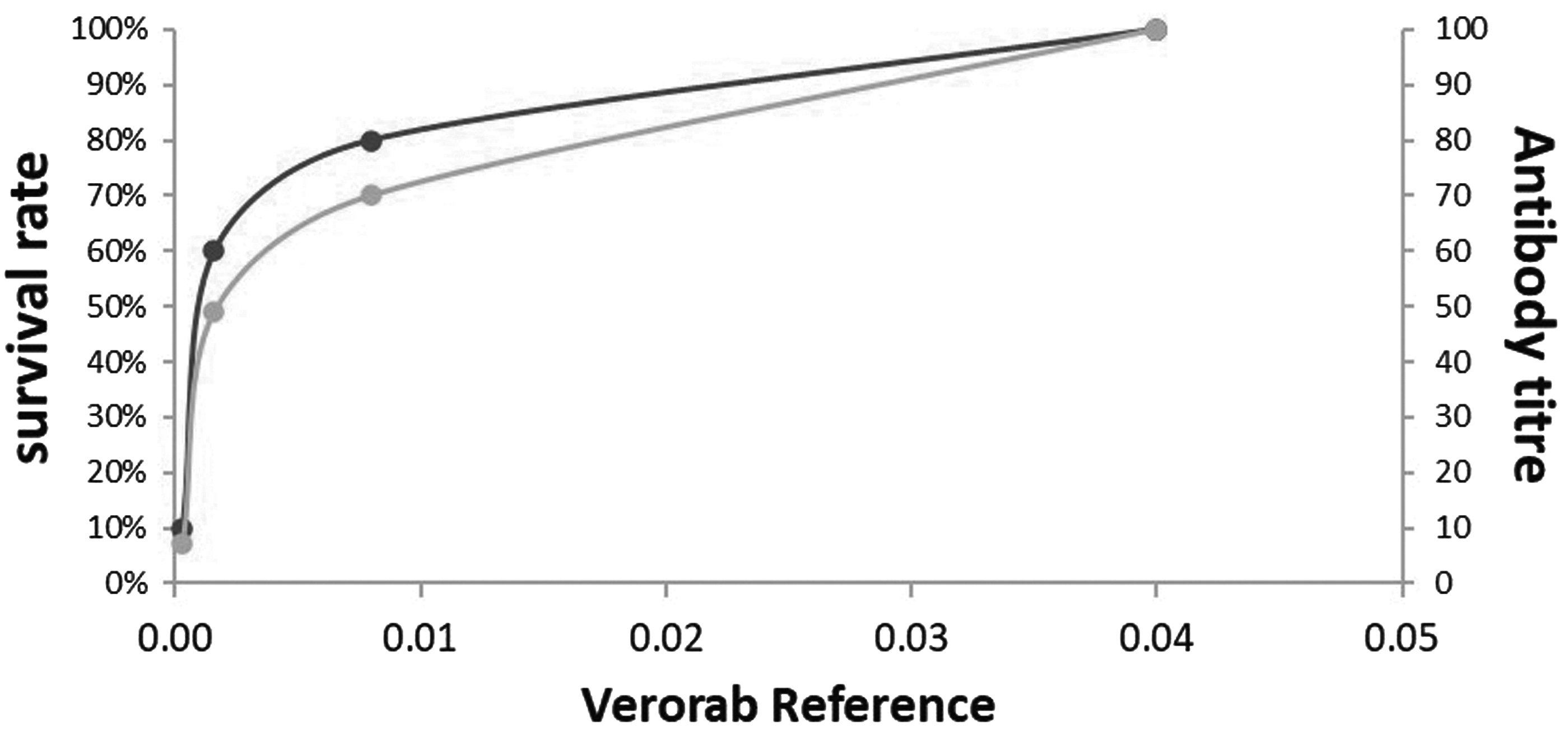
Comparison between survival rate of NIH test and antibody titer of RFFIT for Verorab working Reference standard of rabies vaccine (Sanofi Pasteur, Zuellig Pharma, MIMS, Malaysia). NIH, National Institute of Health.
Potency Evaluation Using Rapid Focus Fluorescent Inhibition Test
ED50%: 50% effective dose; “Berirab,” working standard human rabies immunoglobulin (CSL Behring, Zuellig, MIMS, Philippines).
Effect of ED50% on potency
The ED50% indicates how much of a drug is needed to achieve 50% of the maximum response, and the potency indicates the ability of a vaccine to exert the desired effect in patients. Table 8 showed tested vaccine ED50% compared to that of reference.
The Tested Vaccine ED50% Compared to that of Reference and its Reflection on the Potency
PHA validation and potency evaluation
Validation for PHA is done using same working reference for NIH challenge test and RFFIT “Verorab” (Working Reference standard of Rabies vaccine (Sanofi Pasteur, Zuellig Pharma, MIMS, Malaysia). Validity criteria are met, “PHA titers of duplicate titrations are the same.” The potency of the test as recorded in Table 9 showed good correlation with the reference test NIH.
The Vaccine Potency as Determined by National Institute of Health, Rapid Focus Fluorescent Inhibition Test and Passive Hemagglutination Tests
NIH, National Institute of Health; PHA, passive hemagglutination; RFFIT, rapid focus fluorescent inhibition test; WR, working reference.
Potency correlation of NIH, RFFIT, and PHA tests
The results of the Rabies vaccine potency as determined by NIH, RFFIT, and PHA tests are displayed in Table 9. To decrease variability of tests, the same reference for each experiment in the three tests was used. Therefore, each experiment potency is quite similar. However, all tests pass the acceptance criteria, which are not less than 2.5 IU/mL. Also, we tried to control other conditions by using the same kind of cells, media, and same virus strain with trial to maintain the same conditions for all tests done. Nevertheless, things we could not control included the time of study, so some tests were done in 2015, others in 2016, and the last was done in 2019.
This led to changing the range of potencies between 2.5, 4, 5, and sometimes 6, but in all cases, the samples conformed the acceptance criteria, as recorded in Table 5. The difference between the three tests was the endpoint of each test. In NIH test, which is in vivo, the endpoint was the mice deaths, while in RFFIT and PHA, the endpoint was titer endpoint. This decreases the number of used mice from 200 mice in NIH test to 45 in RFFIT or PHA. It also shortens the time from 28 days in NIH test to 14 days in RFFIT or PHA. Correlation between RFFIT and NIH as well as PHA and NIH using 23 samples of mice sera is displayed in Figures 4 and 5, respectively.

Correlation between the RFFIT and NIH using 23 samples of mice sera.

Correlation between the PHA and NIH using 23 samples of mice sera. PHA, passive hemagglutination.
As shown in Table 10, the three tests (NIH, RFFIT, and PHA) were statistically compared with each other to ensure their validity, using NIH as the reference test by two-way analysis of variance test. F value was 4.1, (p = 0.02). Regression was done for RFFIT and PHA, and the results showed that R squared = 0.83 and 0.39, respectively. The statistical analysis was done using Excel Microsoft worksheet.
Results of Analysis of Variance
Discussion
Rabies vaccine preparations are the only way to prevent rabies infection before the exposure to it, so it is used as prophylaxis (19). Also, it plays a vital role if it is taken as soon as possible after infection exposure (19,20). Its importance comes from the fact that there is no effective treatment for rabies patients already showing symptoms (5). So administration of rabies vaccine (including rabies vaccine in its inactivated form), and rabies immune globulin, is the only approved, effective way for postexposure prophylaxis against rabies in humans (29). The most critical parameter for the quality control of the rabies vaccine is potency (12,17). In Egypt—as in whole authorities—Mouse Challenge NIH test is the main reference test used for potency evaluation (7,10,24).
Many studies are done seeking reduction of animal numbers used in potency tests, or reduction of animal suffering and distress (10,23), or even changing blood collection method from mice (6). This study coincides with all these studies. Nearly 5,000 mice were used in case of NIH challenge test, and about 1,200 mice without challenge were used in case of RFFIT and PHA. Concerning the three tests, it has been found that RFFIT and PHA were highly correlated with NIH challenge test as Pearson correlation was 1, as was determined in this study.
This finding agrees with earlier studies (9,29). This study is divided into four experiments; each experiment has the same condition (reference, animals, and time) to reduce high variability of NIH challenge test, so the potency of each experiment is quite similar. Each experiment consisted of nearly six to eight tests. We calculated the mean, standard deviation, and coefficient of variance of all recorded data.
Potency of all products in the three tests were within acceptance criteria, which is not less than 2.5 IU/dose. We tried to control the most important conditions affecting the respective tests such as kind of cells, culture medi and virus strains. Concerning time, the first two experiments were done in 2015 and 2016 and the last two were done in 2019 and 2020, which led to change in potency range from 2.5 to 6, but all samples conform the acceptance criteria. Dilution is a useful additional check on the accuracy of an assay. It provides information that is of more practical benefit than recovery as it answers the following question: if a sample is diluted, will it give the same result (when corrected for the dilution)—and if not, which is the most accurate result. In NIH, we tried to decrease number of dilutions.
We tried four regimens with one dilution less than the original one. Only one regimen—regimen 2—gave similar results to the original regimen, with 88% (22 samples out of 25). This similarity was attributed to the exclusion of the first dilution, which was the closest to the original one, and the validity criteria were the nearest values to the original as first and second dilutions were similar in values (100–80%). So it was still more than 70%, which was the validity criterion for the first dilution; also, the dilution factor was constant, same as the original one since the dilutions used were the last three and they were sequential. However, in the other three regimens, there were no similar results to the original, except for three samples in regimen 1.
On calculating fold change, we found that in regimen 2, the average fold change was 1 with values between 0.7 and 1.2. The difference between the three tests was the endpoint of each test. In NIH test, the endpoint was the mice deaths, while in RFFIT and PHA, it was the antibody titer. This decreases the number of used mice from 200 mice in NIH test to 45 in RFFIT or PHA; it also shortens the time from 28 days in NIH test to 14 days in RFFIT or PHA.
Regarding validation, before conducting potency test of the three methods, verification was done for them. NIH challenge test is the goldstone test and the main test for potency of rabies vaccine preparations since 1950 (6,23). So verification was done by comparing three different references: NIBSC, National reference of China, and Verorab as working reference standard of rabies vaccine for this study. The three references gave the same ED50%, 2.7, but with different potencies, 4.0, 3.3, and 4.5, respectively “all within acceptable limit.” This difference was due to number of animals used. On the other hand, RFFIT test is also a WHO standard assay (q).
So we analyzed the three tests regarding the selective validation parameters, including “sensitivity, specificity, repeatability, and coefficient of variance,” and it was done statistically, and their results were found to be within acceptable limits. The last method “PHA” is also a WHO-described method, and validity criteria were checked during the test.
In literature, many comparisons had been done such as comparison between mouse neutralization test and RIFFT (4,21) or comparison between NIH challenge test and ELISA (3,6). Some reports studied each method in detail, such as studying the effect of each factor on PHA method (11) like erythrocyte concentration and the pH. Others studied evaluation of immune response to rabies vaccine (2). However, our study is the first comparative study between the three methods concerning potency test for rabies vaccine preparations. It is a comprehensive study, starting with validation of the three methods, moving to testing real samples with three methods under almost similar conditions, followed by statistical analysis of the obtained data to ensure accuracy and validity of this study.
Finally, from this study, we can conclude that (i) both RFFIT test and PHA tests can be used for the potency evaluation of Rabies vaccine products indicated for human use. However, caution has to be taken for both tests during assays and validation and should be preconducted, (ii) both methods can be efficiently utilized for calculations of ED50% for rabies vaccine preparations, (iii) also in the most tested rabies vaccine preparations, the results of both RFFIT and NIH tests were comparable, while PHA still needed further studies, (iv) both RFFIT and NIH tests have to be carried out to complement and reinforce each other as they provide a comprehensive picture of the product potency, and (v) RFFIT test is more accurate, fast, and sensitive than NIH, while PHA is not as accurate as RFFIT.
However, it still gives reliable results and is easier, faster, and cheaper than RFFIT. On the other hand, NIH test has limitation, including its high variability and consumption of large numbers of animals.
Footnotes
Acknowledgments
The authors would like to thank and express their gratitude to all members in Viral Control Unit and Animal house at Central Administration of control of Biologicals and innovative products and clinical studies, Egyptian Drug Authority, Egypt, for providing facilities that allowed this work to be achieved.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.