
Abstract
Zika virus (ZIKV) infections have gained notoriety due to congenital abnormalities. Pregnant women have a greater risk of ZIKV infection and consequent transmission to their progeny due to the immunological changes associated with pregnancy. ZIKV has been detected in amniotic fluid, as well as in fetal and neonatal tissues of infected pregnant women. However, the mechanism by which ZIKV reaches the fetus is not well understood. The four dengue virus serotypes have been the most widely used flaviviruses to elucidate the host–cell entry pathways. Nevertheless, it is of increasing interest to understand the specific interaction between ZIKV and the host cell, especially in the gestation period. Herein, the authors describe the mechanisms of prenatal vertical infection of ZIKV based on results from in vitro, in vivo, and ex vivo studies, including murine models and nonhuman primates. It also includes up-to-date knowledge from ex vivo and natural infections in pregnant women explaining the vertical transmission along four tracks: transplacental, paracellular, transcytosis mediated by extracellular vesicles, and paraplacental route and the antibody-dependent enhancement process. A global understanding of the diverse pathways used by ZIKV to cross the placental barrier and access the fetus, along with a better comprehension of the pathogenesis of ZIKV in pregnant females, may constitute a fundamental role in the design of antiviral drugs to reduce congenital disabilities associated with ZIKV.
Introduction
Zika virus (
Approximately 80% of ZIKV infections are estimated to be asymptomatic, the remaining 20% develop a febrile illness with mild symptoms that last from 2 to 7 days (44,71,73) (Fig. 1), presenting in many cases a maculopapular rash, conjunctivitis, muscle aches, joint pain, malaise, and headache (56,104). In some adults, ZIKV can cause serious complications, such as multiorgan failure, meningitis, encephalitis, thrombocytopenia, and Guillain–Barre syndrome (76). In pregnancy, ZIKV has been associated with spontaneous abortion, intrauterine growth restriction (IUGR) (66), intracranial calcifications, development of microcephaly, congenital zika syndrome (CZS) (16,100), and other abnormalities of fetal development (61,72,76).
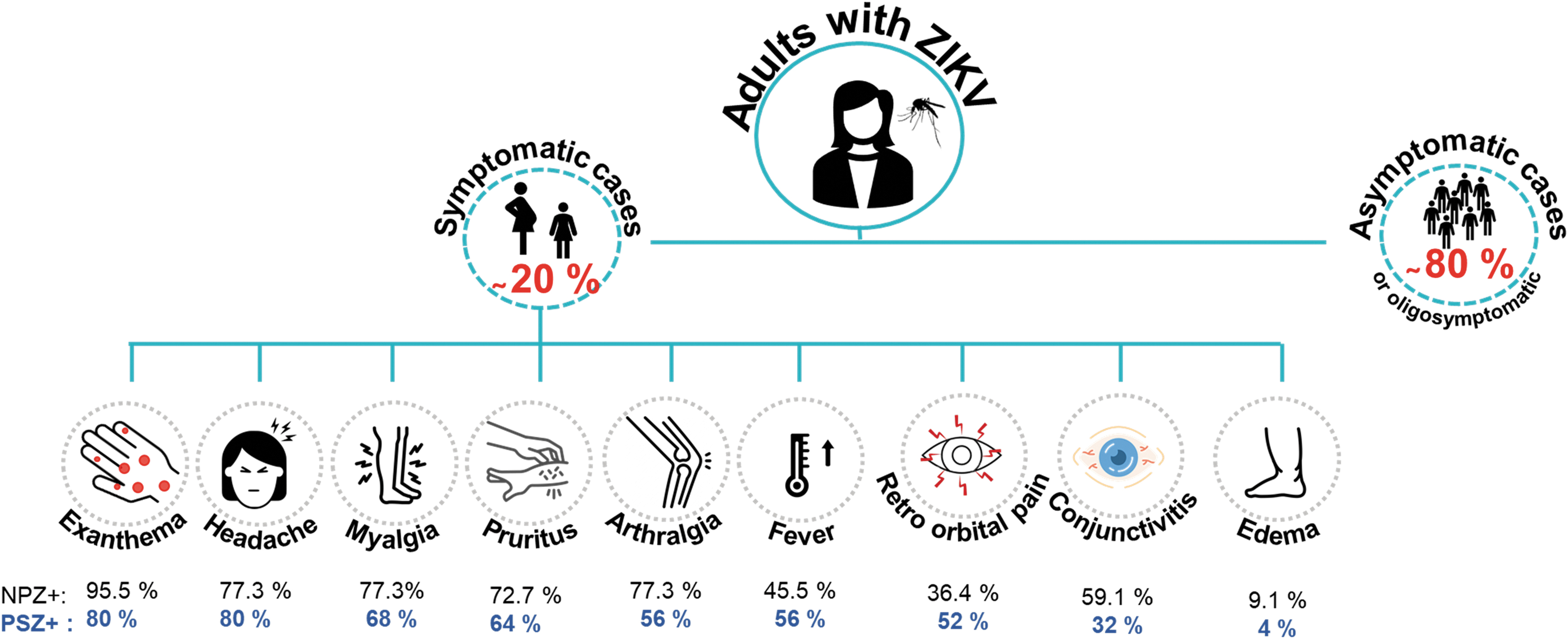
Clinical manifestations of ZIKV infection in pregnant women symptomatic ZIKV positive (PSZ+) and Nonpregnant women ZIKV positive (NPZ+). ZIKV, Zika virus.
Congenital disabilities associated with ZIKV often occur when infections arise in the first trimester (46), mainly between 8 and 16 weeks of gestation (WG) (56,93). Even in pregnancies in which the baby is born without visible signs of infection, there is the possibility of him or her developing postnatal microcephaly, and atrial and ophthalmic anomalies (35).
Although the mechanism through which ZIKV reaches the fetus is not well known, it is understood that infected monocytes in the bloodstream allow the virus to gradually spread to different tissues and organs (37). Some researchers have detected viral RNA in several tissues, such as the male and female genital tracts, placenta, and decidual tissues (56,93). It has also been detected in amniotic fluid and fetal and neonatal brains (10). Additionally, IgG antibodies (Abs) against ZIKV, have been detected in newborns (10,56,89,94). Therefore, understanding the mechanisms of prenatal vertical infection is essential to prevent the teratogenic effects of ZIKV infection.
The transplacental route was previously summarized by Cao et al., describing the scientific evidence available up to 2017, including trials in murine models (11). Nevertheless, to strengthen this information, the review herein discusses the most recent advances supporting this possible pathway along two tracks, paracellular and transcytosis mediated by extracellular vesicle (EV) pathways. It includes evidence obtained from nonhuman primate models, as well as explanations of the paraplacental pathways and the antibody-dependent enhancement (ADE) phenomenon as potential ZIKV transmission routes. The immunological changes in maternal and placental tissues that affect ZIKV replication throughout gestation and the complications associated with ZIKV crossing the placenta are also considered.
Placental Cells Involved in Viral Infections
The human placenta is the main barrier between the fetal and maternal compartments during pregnancy. It comprises trophoblasts derived from gestation and maternal tissues that exchange gases, nutrients, and immune effectors of both innate and adaptive responses (4). Over the first WG at the maternal–fetal interface, the trophoblastic cells differentiate into anchoring villi, formed by invasive cytotrophoblasts (CTBs) that anchor to the uterine tissue and into floating villi, composed of extravillous cytotrophoblasts (EVCTs) and syncytiotrophoblasts (STBs), which are in direct contact with maternal blood (25). The remarkable selectivity of these barriers is mainly due to STB, and a multinucleated tissue consisting of fused layers that border the chorionic villi.
The core of the villous structure beneath the STB layer, consists of CTB, Hofbauer cells (HBCs), fibroblasts, and endothelial cells. The maternal–fetal interactions take place in the placental villous structures that contact the maternal blood and the adjacent decidua. Since STB are in direct contact with maternal blood, they are also in direct contact with the circulating pathogens.
STB cells have defense mechanisms against infections (86,111). As part of the antiviral mechanism, they secrete type III interferon (IFN-λ) (6,48) and become increasingly resistant to viral infections toward the end of pregnancy. These cells do not express genes encoding most attachment factors implicated in ZIKV entry, but express many genes associated with antiviral defense. In this sense, maternal immune system plays an essential role in the protection against pathogenic invasion and Abs transfer from the mother to the fetus. Therefore, STB constitutes a barrier against circulating pathogens. Also, Abs transfer depends on STB through the neonatal Fc receptor (FcRn) (92). Pathogens must cross the STB layer in the floating villi and infect the EVCT to internalize into the villous nucleus and the placenta.
However, it has been widely reported that HBCs can harbor viruses and serve as reservoirs within the placenta. These cells may contribute to transmitting several pathogens to the fetus (85). According to Jurado et al., HBCs and primary placental-specific fibroblasts are permissive cells for the infection and replication of ZIKV. Only HBCs demonstrated susceptibility ex vivo in the context of placental tissues (49,90). The resulting infection in the placenta elicits the proliferation and hyperplasia of HBCs in the chorionic villi, which represents the primary placental response during a transplacental ZIKV infection, in the second and third pregnancy trimesters (49,85,87,90).
Nevertheless, these findings could not be compared with in vivo conditions, because HBCs may promote specialized cell subsets in response to their microenvironment (85), including several hormones (110). In agreement with this, STB as a maternal–fetal layer, could indirectly mediate the infection of HBCs and explain this effect through the ADE. This mechanism has been proposed as responsible for some pathogens, such as cytomegalovirus, avoiding the placental humoral responses (55).
Antibody-Dependent Enhancement
The ADE is a phenomenon that may occur when subneutralizing Abs crossreact with a related virus, or when crossreactive Abs without neutralizing nature bind to epitopes not involved in cell attachment and viral entry (9,29,50). Consequently, neutralization decreases and the viral entry into the new host cells expressing Fc-gamma receptors (FcγR) increases (51,98,99).
It is known that whole IgG molecules or crystallizable fractions (Fc) can be transferred to the neonatal circulation by the FcRn of the STB (74), therefore, placental transfer of Abs represents a possible pathway for virus–Abs complexes to reach and infect the fetus. A current hypothesis suggests that the ZIKV ADE is due to previous exposure to closely related viruses (94).
ADE has been evaluated among different DENV serotypes, in an attempt to explain dengue hemorrhagic fever, caused by a second DENV serotype infection, where an increase in viremia and a greater risk of developing severe disease were described. This correlation was studied among other flaviviruses, such as ZIKV, due to the phylogenetic relationship and structural similarities with DENV (8,12,28,45,75,97). Garg et al. reported the intensification of ZIKV infection by Abs, in symptomatic and asymptomatic WNV-seropositive (WNV+) individuals. The authors have proven that WNV+ sera contain Abs that exhibit neutralizing activity for WNV but, in turn, have low neutralizing action against ZIKV (39).
Furthermore, they indicated the association of ADE with the presence of Abs against the envelope protein of Flavivirus (48). Fowler et al. reported this phenomenon in offspring of mice whose Abs against ZIKV were transferred by ZIKV-immune mothers, facilitating DENV infection and increased the risk of severe dengue in the pups (38). Conversely, it has been confirmed that Abs in sera from subjects infected with DEN are capable of binding to ZIKV but with a poor neutralization effect, confirming an ADE effect in cell culture, with human primary monocytes, in which FcγRI, FcγRII, and FcγRIII receptors demonstrated an essential role in increasing ZIKV infection (53).
Some researchers have studied the flavivirus vertical transmission in the context of a possible ADE phenomenon, comparing pregnant mice with previous flavivirus infection with naive pregnant mice. Both groups were subsequently infected. Rathore et al. compared DENV-naive pregnant mice with DENV-immune pregnant mice after ZIKV exposure. They demonstrated that anti-DENV Abs stimulated ZIKV infection in fetuses of DENV-immune mice in contrast with naive pregnant mice. They also found FcRn to be present in placenta cells, before and during the detection point of the ZIKV infection in embryos. Additionally, the maternal ZIKV infection increased the FcRn expression on endothelial cells and STB (82).
Brown et al. demonstrated that pre-existing Abs against DENV led to an intensification in the pathogenesis of ZIKV in Stat-2−/− pregnant mice, leading to increased placental damage, growth restriction, and fetal resorption, compared with those mice without previous DENV infection (8).
These findings align with the fact that the presence of anti-DENV Abs enhances ZIKV infection in human CTB and HBCs, likely leading to an inflammatory environment on the placenta. An increase in the number of infected trophoblasts compromised the differentiation and development of chorionic villi and uterine invasion (10,12,75,97). They also suggest that maternal Abs boost transplacental infection of mice fetuses resulting in an aggravated phenotype compatible with microcephaly (82). These results support the fact that primary infection with closely related flaviviruses could increase the severity of ZIKV infection in pregnant women and increase the probability of CZS.
Nonhuman primates represent a more human-like model for infection and fetal development; therefore, these models have been used to study the effect of ZIKV infection. Few studies provide information about the ADE effect in these models, nevertheless, the capacity for fetal infection has been confirmed (68). Viral infection assays carried out during the first and second pregnancy trimesters show that chorionic villus preferential viral replication remains in these animals, which could point to CTB. Although ZIKV infection induces neutralizing Abs responses even when they are considered protective, after ZIKV infection in DENV-immune primates, significantly higher viral RNA loads were found in the chorionic villi maternal–fetal interface compared with nave primates (23,24).
While low levels of crossreactive ZIKV Abs were found at the time of challenge in all DENV-immune primates, in some cases, these titers correspond to those reported to increase the risk of severe DENV in humans (1:80). Unfortunately, the study did not include the analysis of placental tissues during pregnancy, therefore, the role of crossreactive Abs and viral infection in these primate tissues remains to be explored (24). The relevance of studying the Ab–receptor interactions is highlighted by the fact that passive administration of ZIKV-neutralizing Abs with an Fc-modified region that could prevent the binding to FcR, reduces the maternal viremia and limits vertical transmission (101).
However, the cases in which ZIKV can access placental tissues and convert them into a virus reservoir, suggests that the mechanisms by which the virus accesses the maternal–fetal interface may be varied. Furthermore, passive maternal Abs transfer could decrease ZIKV vRNA in amniotic fluid and other fetal tissues during the third trimester (19), promoting viral clearance and thus making detection at delivery difficult. At the same time, the maternal–fetal interface acts as a reservoir, contributing to prolonged viremia, which is associated with accentuated visual orientation skill deficits in infants with DENV/ZIKV exposure compared with ZIKV exposure only (5).
According to literature, maternal Abs transportation through the human placenta begins approximately at 12 WG. The Abs levels in fetal blood remain low until the second trimester, when they gradually increase until the end of gestation, due to the expansion of the STBs, which expand the contact area for the transfer and higher expression of FcRn. It was reported that around the second trimester, Abs levels are 10% of the maternal concentration and 50% between 28 and 32 WG, at the 40 WG they reach 20–30% more than the maternal levels (74,92,106).
Despite the low levels of FcRn expression during early gestation, these may mediate the transfer of IgGs enabling STB to promote the transcytosis of ZIKV–Abs complexes, toward the fetal side of the placenta. The possible presence of crossreactive Abs and low Abs levels transferred from the mother to the fetus in early pregnancy, could limit effective neutralization and enhance infection of other placental cells.
In this case, CTB infection would be favored in the second trimester of pregnancy, damaging fetal development, explaining the prevalence of congenital disabilities, and the risks of abortion associated with ZIKV infections in the early stages of pregnancy (8,10,30). Similarly; the binding of Abs to ZIKV-related viruses (such as DENV), could provide an entry pathway through the STB, to subsequently interact with the FcγRs present in the HBCs promoting viral entry, especially in the presence of IgG1 and IgG3. This is consistent with the IgG response in subjects with symptomatic dengue and with the affinity of the FcRn for the transfer of the different IgG subclasses (IgG1> IgG4> IgG3> IgG2) (74,106,113) (Fig. 2).
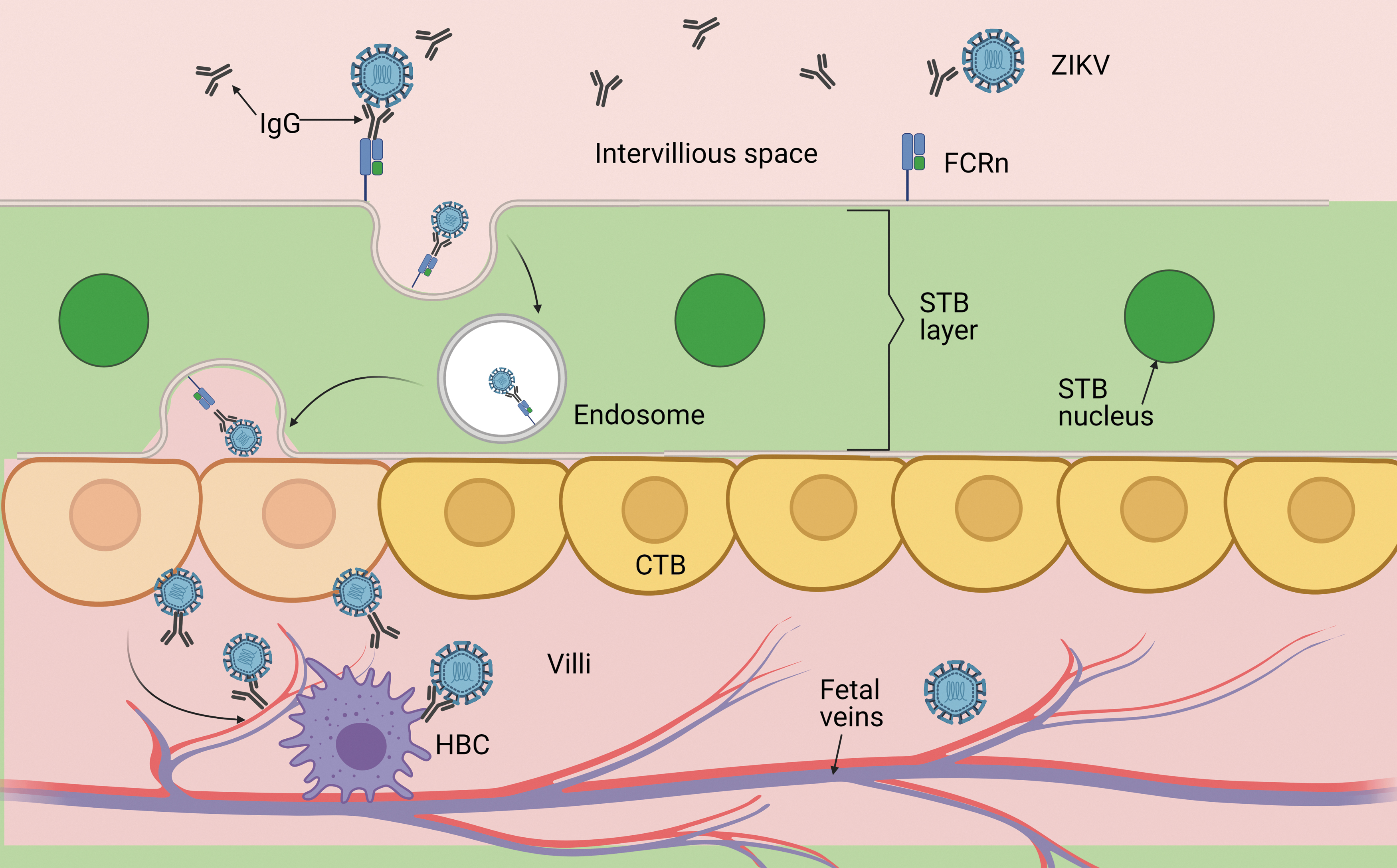
ZIKV transmission due to ADE. Current evidence suggests that subneutralizing existing Abs could bind to ZIKV, followed by internalization through the FcRn in STB, to infect CTB and HBCs, further spreading the ZIKA viral infection to the fetus. Abs, antibodies; ADE, antibody-dependent enhancement; CTB, cytotrophoblast; FcRn, neonatal Fc receptor; HBCs, Hofbauer cells; STB, syncytiotrophoblasts.
Considering that the ADE phenomenon includes the binding of Abs to epitopes that are not involved in viral entry, an additional hypothesis could be the interaction of non-neutralized flavivirus with a variety of receptors in placental and fetal cells that could lead to inflammatory symptoms, resulting in placental and neurological damage, depending on the set of cells affected (Table 1). Viral RNA has been detected before IgG transference to the fetus, and it has been proposed that ZIKV could reach the fetus through other infection mechanisms (2). The degree of ZIKV maturation depends on the cell where virions are assembled. As a result, a diversity of particles allows for different receptors and pathways to the new cell host (Table 1) (83,84,95).
Potential Entry Receptors for Zika Virus
Transplacental Infection
There are reports where different cell types isolated from placentas are permissive to ZIKV infection, such as macrophages, endothelial cells, amniotic epithelial cells (AmEpC), trophoblast progenitor cells (TBPC), human umbilical vein endotelial cells (HUVEC), human placental fibroblasts (HPF), and primary placental CTBs. Once these cells are infected, the placental proliferation process stops (64,79), explaining the IUGR and fetal death (65). Aagaard et al. reported that primary human placental trophoblasts are permissive for ZIKV replication and exhibit transcription of four receptors for ZIKV entry: AXL, DC-SIGN, TYRO3, and TIM-1 (1).
ZIKV crosses the placental barrier modulating the membrane physical breakdown, making it permeable and destroying the placental barrier integrity. The nonstructural protein (NS1) of several flaviviruses (ZIKV, DENV, WNV, JEV, and YFV) is responsible for immune evasion and vascular permeability (77,81). In vitro, NS1 increases the permeability of endothelial cells of the umbilical vein and brain, among others, causing vascular leakage and potentially influencing the spread and pathogenesis of the virus (54,77,78).
NS1 alters glycosaminoglycans from glycocalyx in endothelial cells, causing the anchoring villi's permeability in early gestation (40,78,103), due to the liberation of hyaluronic acid and heparan sulfate in stromal fibroblasts, HBCs, and trophoblasts, by production of the enzyme hyaluronidase (94). These findings are key to demonstrating placental transfer, illustrating the entry of ZIKV in the fetal compartment (Fig. 3).
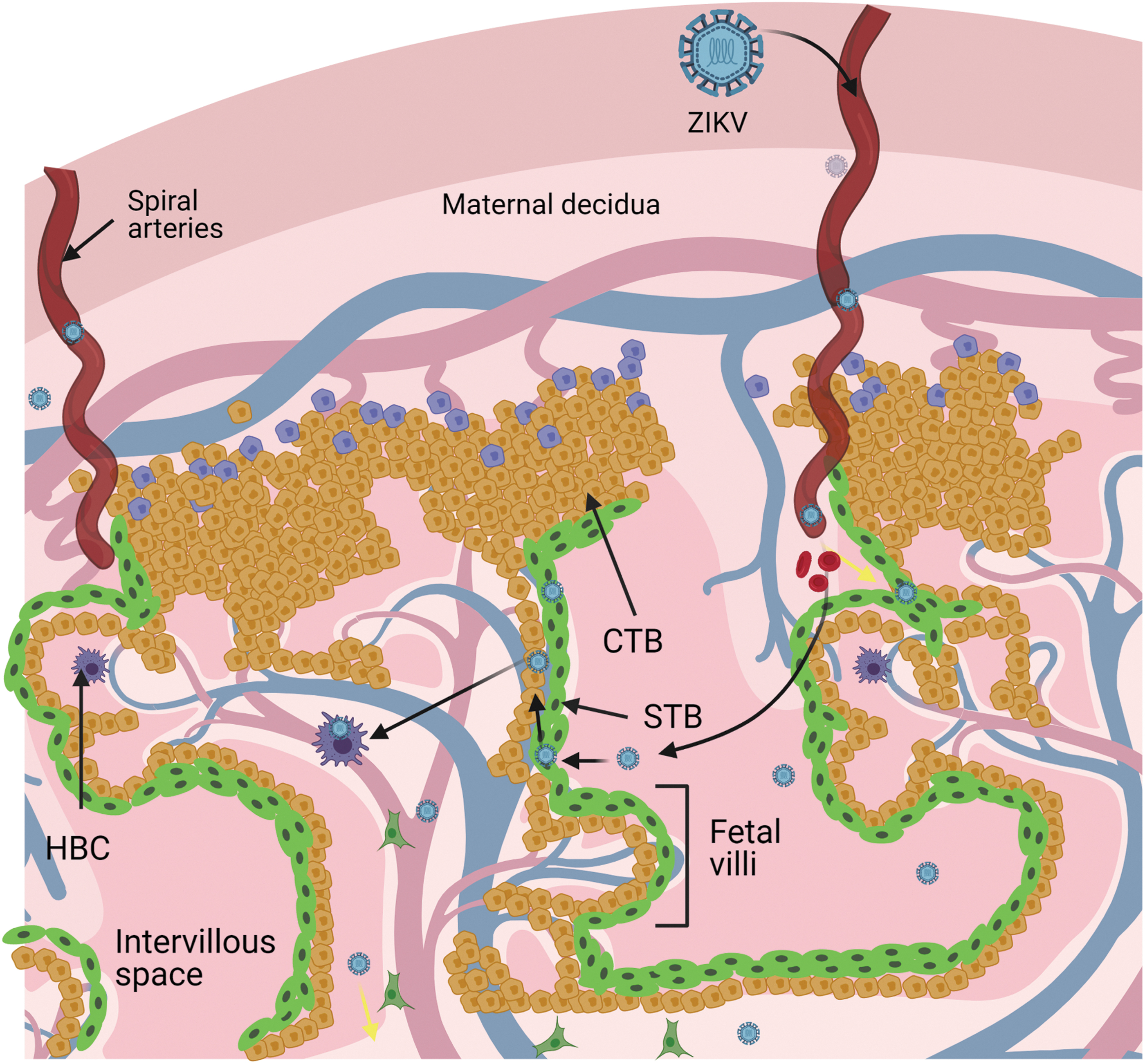
Model of transplacental infection according to Tabata et al. During early pregnancy, the trophoblastic cells that form the chorionic villi are susceptible to ZIKV infection. Black arrows indicate the path that ZIKV takes to internalize into chorionic villi.
Paracellular Pathway
Some recent investigations argue that ZIKV could cross the placental barrier through the paracellular pathway (between cells) (14,72). Epithelial tissue cells are connected to one another by tight junctions (TJs) on the apical and basolateral surface of cells, regulating the transit through epithelial tissue. TJs are composed of various proteins, such as claudins, occludins, and junctional adhesion molecules (JAMs) (33,58).
In STB, these TJs and their function as paracellular seals have been described in several studies, reporting different proteins such as occludin, claudins (1,3,4, and 16), and zonula occludens protein (ZO-1). ZIKV infection causes leakage of the placental barrier in CTB and STB cells, by altering junction barrier integrity, due to decreased amounts of essential binding proteins ZO-1, occludin, and claudin-4 (15,17,60). This suggests that ZIKV can open the paracellular pathway modifying placental TJ's composition and increasing paracellular permeability (Fig. 4).
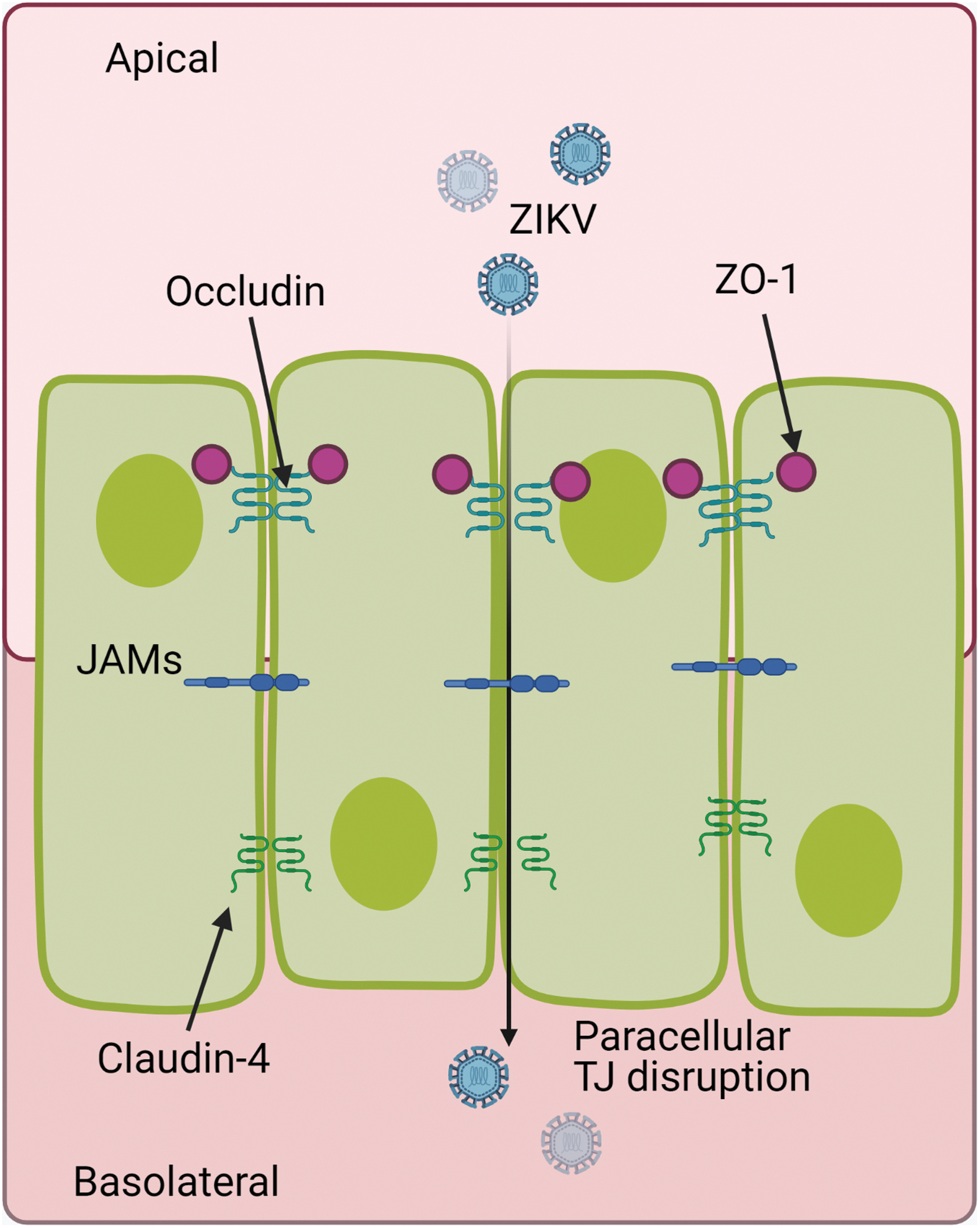
Model of paracellular infection by ZIKV. The virus may invade the placenta through paracellular entry, following the disruption of junctions between STB. JAMs, junctional adhesion molecules; TJ, tight junctions; ZO-1, zonula occludens protein.
These findings are supported by experiments on primary human brain microvascular endothelial cells. Leda et al. found that occludin and claudin-5 levels are downregulated by three different ZIKV strains, concluding that ZIKV could cause alterations of TJ protein expression, in a strain-dependent manner (52). Unfortunately, this TJ modification depending on the virus strain has not been confirmed on the placental barrier.
It is well documented that ZIKV changes the cytoskeleton organization and host cell metabolism (18,21,22,109). Due to this fact, the actin skeleton's role in ZIKV infection has been explored, demonstrating that the interaction between the envelope (E) protein of ZIKV and F-actin leads to the reorganization of the actin filament, thereby compromising the blood–testis barrier (BTB) integrity (69).
Additionally, the viral NS1 protein has been associated with an increase of matrix metalloproteinase 9 (MMP9) levels, which also led to a significant degradation of TJ proteins and collagen type IV, essential in BTB maintenance, thus, inducing vascular leakage by altering endothelial cell adhesion, and leading to the hyperpermeability of BTB (47,75). These findings offer a new ZIKV entry mechanism to the blood–tissue barrier (69); however, this placental barrier assumption must be further studied.
Transcytosis Mediated by EV Pathway
Additionally, EVs are considered signaling mechanisms between cells and therefore have been proposed as an explanation for another viral dissemination route (20). In the case of flaviviruses, this mechanism has been evaluated with growing interest. The EVs are essential in the crosstalk between mother and fetus, in processes such as implantation, trophoblast invasion, and proliferation (26).
Although, few studies have focused on the effect of maternal EVs on the fetus, it is well known that EVs can cross the placental barrier and deliver molecules (91). Furthermore, the EVs share similar characteristics with viruses: structure, size, lipid composition, and uptake (107). Several scientists have proposed the microvesicles as a transplacental ZIKV route in human pregnancy, by hijacking exosome-mediated cell–cell communication (110). York et al. demonstrated that ZIKV-infected cells secrete distinct EV subpopulations, containing vRNA and are infectious to noninfected cells (108).
They concluded that ZIKV uses CD63 tetraspanin during the replication stage for exocytosis of new virions and it is probably involved in genomic RNA packaging due to an increase of CD63 expression rates on infected cells compared with noninfected cells, which were observed. It has been reported that tetraspanins are essential for specific steps in the entry and exit of several viruses such as human papillomaviruses, hepatitis C virus, influenza A virus, Middle East respiratory syndrome coronavirus, and others (31,36,41,67). Finally, Chen et al. demonstrated the presence of infectious virions packaged in EVs (surrounded by a bilayer lipid membrane 300–500 nm in diameter) from infected placentas of ZIKV-positive donors (13).
Zhou et al. reported high levels of viral proteins and vRNA content in exosomes derived from neuronal cells. Their findings suggest that exosome-mediated dissemination is faster than mature virion-receptor dissemination (112). Martínez-Rojas et al., found that the EV produced by ZIKV-infected mosquito cells contain E protein and vRNA, and have infective capacity. They also reported that this EVs stimulate the activation and differentiation of naive human monocytes, inciting an inflammatory response with increased endothelial permeability (57).
In another approach, ZIKV-infected hcMEC/D3 EVs were evaluated, where Fikatas et al. observed that both EVs and ZIKV manage to generate changes in capacitance, suggesting that both are capable of temporarily altering the integrity of the monolayer culture, by alteration of vascular endothelial cadherin, mediated by EVs as a mechanism of ZIKV transmission in the blood–brain barrier system (34). These findings point to a vertical transmission mechanism in which the ZIKV hijacks the exosome pathway on mother cells, to cross the placental barrier mediated by the exchange of EV between the mother and the fetus, to enter fetal circulation (Fig. 5).
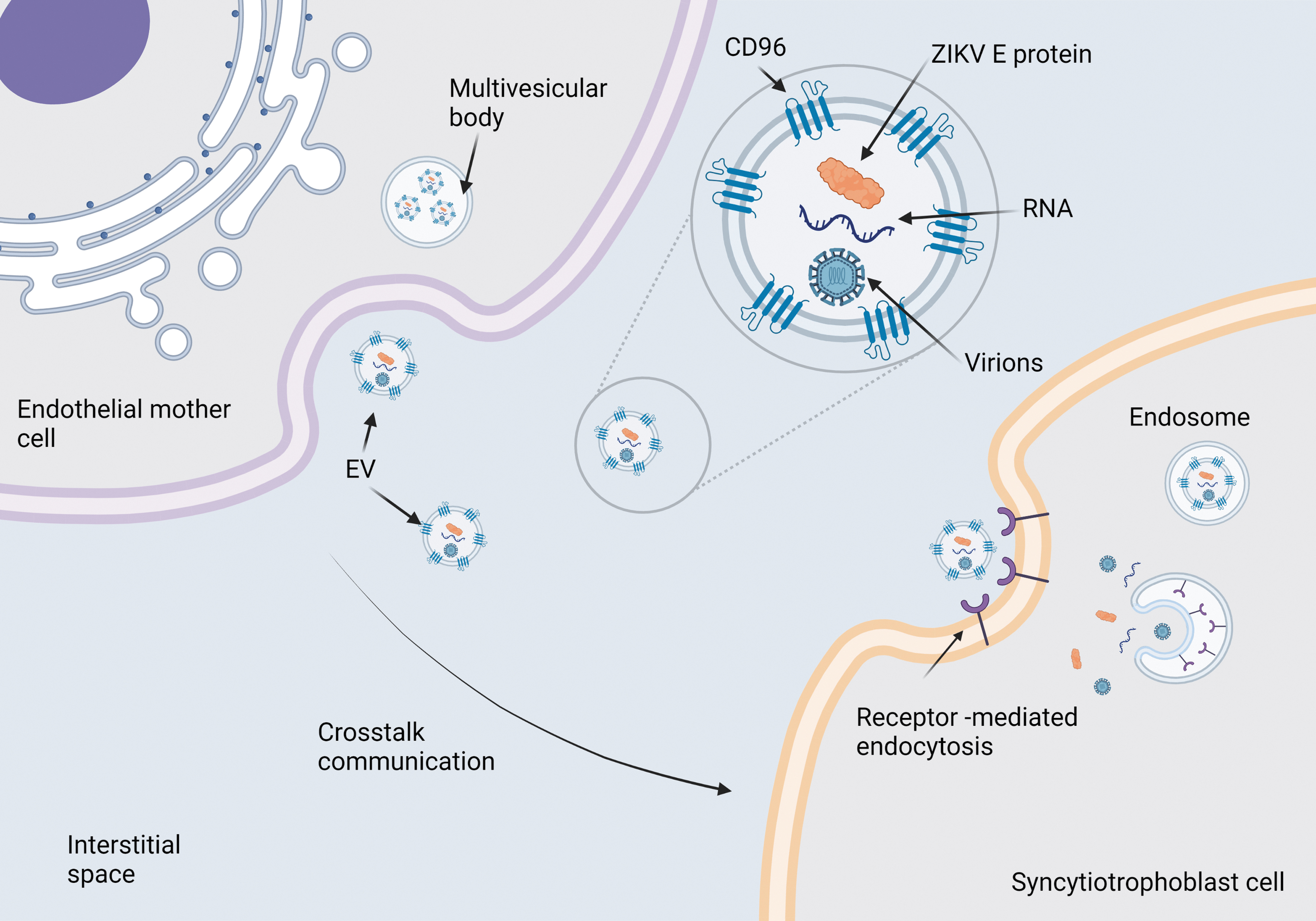
Model of transcytosis mediated by EV pathway. EVs can carry on viral proteins, RNA or viruses and infect to STB cells to cross barrier placental. EVs, extracellular vesicles.
Paraplacental Transmission
There is strong evidence that the human placenta provides the main barrier to virus transmission, as it matures defense mechanisms such as production of cytokines, such as IFN-I (IFN-α, IFN-β), IFN-II (INF-γ), and IFN-III, helps trophoblast cells become less vulnerable to ZIKV infection by the second trimester of gestation (95). To explain cases in which pregnant women whose illnesses took place during the second and third trimesters of pregnancy, Tabata et al. proposed a paraplacental route (70,96), where the decidua parietalis is in contact with the chorionic membrane, from the 15 WG. It covers the entire surface as the pregnancy progresses. Therefore, ZIKV could spread from the decidua parietalis to the amniochorionic membranes.
The detection of ZIKV in amniotic fluid and AmEpC supports this potential pathway (10,59,96) due to the viral circulation in the amniotic fluid, which could infect susceptible epithelial cells in the fetus (Fig. 6) (55). Weisblum et al. demonstrated viral infection spread in the maternal decidual tissues during gestation. The trophoblast chorionic villi derived from the fetus showed a downward replication of ZIKV with the advance in gestational stage (105).
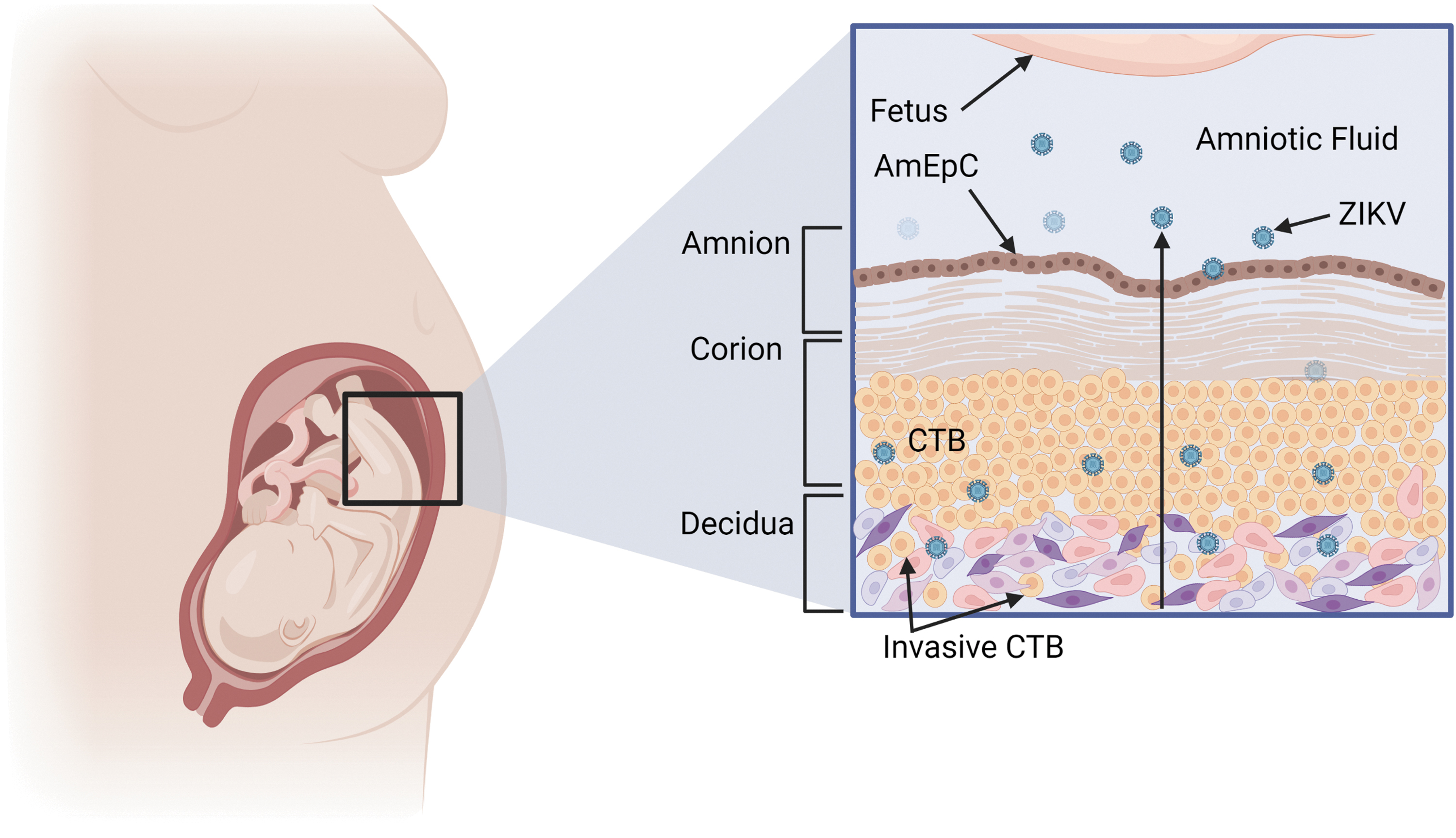
Model of paraplacental infection according to Tabata et al. (96). In mid-gestation, the placenta comes in contact with the parietal decidua's uterine wall, and the invasive CTBs form new anchoring villi. ZIKV infects the parietal decidua cells to the villi, comes in contact with AmEpc, and follows the amniotic fluid toward the fetal epithelial cells. AmEpC, amniotic epithelial cells.
Consistent with these results, Guzeloglu-Kayisli et al. outlined that human decidual cells act as viral reservoirs and have a role in placenta–fetus viral infections in the first and second trimesters, which explains the vulnerability and fatal consequences observed in infections in the early stage and adverse effects at the late stage of pregnancy (43). Moström et al. led a study in NHP to identify the immune profile of the maternal–fetal interface in healthy conditions and its alteration by ZIKV infection, demonstrating a reduced activation and cytotoxicity of decidual memory T lymphocytes, and decreased CXCRE receptor expression in ZIKV infection. These results indicate local immunosuppression and impaired immune recruitment, proposed by the authors as possible mechanisms of vertical transmission (62).
Implications in Viral Persistence
Little has been described about human placenta metabolism alterations caused by the ZIKV prolonged infection. Santos et al. described morphological and molecular changes in the infected placenta. They found overexpression of the apoptosis inhibitor BCL-2 in STB of late pregnancy and a decrease in the expression of human chorionic gonadotropin compared with noninfected cells (89).
ZIKV has also been shown to downregulate the anti-inflammatory protein annexin A1 (ANXA1) in infected placentas. These results are consistent with other studies that demonstrate inflammatory cell and cytokine recruitment at the maternal–fetal interface (80). However, ZIKV infection affects placental function due to impaired cell migration at the maternal–fetal interface (a critical process for uterine spiral artery remodeling) and overexpression of GLUT-3, resulting in greater glucose use, and consequently, prolonged latency of the infection (102).
Conclusion
ZIKV infection in early pregnancy causes severe fetal defects designated as CZS, which include microcephaly, neurological impairment, cerebral calcification, retinal damage, and others. However, ZIKV infections in late pregnancy are associated with adverse perinatal outcomes, including IUGR, fetal loss, and others. While the mechanisms of vertical transmission from mother to fetus have been well studied, these and other effects have not been entirely elucidated. For example, it is unclear what drives disease severity in each individual and how it is possible for an asymptomatic mother to have a severely affected fetus or fetal loss.
There is an increasing need to identify these mechanisms to contribute to the design of vaccine candidates and antiviral drugs. It is essential to consider pregnant women as a vulnerable group to prevent the dissemination of ZIKV and its secondary effects. This review helps to advance the comprehension of different ZIKV infection pathways in the placental barriers, based on the experimental evidence during infection, in both, in vitro and in vivo gestation animal models, as well as ex vivo assays and natural infections in pregnant women.
In this study, were described four possible routes of ZIKV fetal infection based on placental approaches, such as ADE effect, transcytosis mediated by EVs, and paracellular and paraplacental pathways, to explain why pregnant women are susceptible to ZIKV infection and why the damages are more insidious during first trimester than in late pregnancy.
The trophoblast cells of the early developing placenta and endothelial cells of the mother express many attachment receptors that facilitate ZIKV entry to the host cell, in contrast to late and terminal placental cells. Both types of permissive cells in the early pregnancy are more susceptible to placental infection. They would explain the increase in pregnancy complications at this stage, where the trophoblast is differentiating into CTB and STB, and IgG transfer is low.
Furthermore, ZIKV induces placental inflammatory response due to recruitment of proinflammatory cells and cytokines in the maternal–fetal interface and overexpression of antiapoptotic regulator BCL-2, which allows persisting infection during the whole pregnancy. ZIKV infection also alters the composition of placental TJs between cells. It increases paracellular permeability mainly due to a reduction in occludin-4 expression in endothelial tissue; therefore, this may significantly impact the paracellular transit through the placenta to the fetus.
Considering that STBs are involved in the transfer of virus–Abs complexes, this access pathway for viral particles to permissive cells becomes especially relevant from the second trimester of pregnancy, where increased expression of FcRn could favor the ADE effect in different cells, such as CTBs, HBCs, glial, and neural cells in the mid-gestation period. Consequently, in the follow-up of pregnancy, it is relevant to assess the period elapsed since a previous exposure to flaviviruses other than ZIKV due to nonspecific or subneutralizing Abs levels play an essential role in ADE.
As mentioned, ZIKV has a broad tropism and the capacity to cross cellular barriers such as placenta. EVs have been reported to cross the placental barrier and we hypothesized ZIKV utilizes different biogenesis pathways in cells for the production of empaqueted EVs with proteins and infective RNA. Consequently, it is possible that the ZIKV can secrete infective EVs to facilitate cell to cell infection into the placental barrier. Finally, maternal ZIKV infection compromises the placental barrier by infecting fetal trophoblasts, thereby entering the fetal circulation imparing development, producing adverse perinatal outcomes, depending on the stage of pregnancy when the infection occurs.
Footnotes
Acknowledgment
E.V.-S., M.B.-F., and L.Z.-C. acknowledge the individual fellowship granted by CONACYT during the PhD. All images were created with BioRender.com.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported with funds provided by the “Fondo de Cooperación Internacional en Ciencia y Tecnología” CONACYT-BMBF 2016 Grant: FONCICYT-291205.