
Abstract
The global pandemic of SARS-CoV-2 in the past 2 years has aroused great attention to infectious diseases, and emerging virus outbreaks have brought huge challenges to the global health system. Viruses are specific pathogens that completely rely on host cells for their own survival and disease transmission. At present, a growing number of studies have proved that inducing the death of virus-infected cells can prevent the spread of virus and promote disease recovery. Therefore, many ways to induce the death of infected cells are considered to be beneficial to host immunity. Cell death is a basic biological phenomenon. Programmed cell death (PCD), as an important part of the host's innate immune response, provides effective protection against virus transmission. Pyroptosis, apoptosis, and necroptosis are the most commonly studied pathways of PCD. Recent studies have found that three pathways of cell death can be activated during virus infection. More and more studies have shown the existence of extensive connections between PCDs, and this complex relationship is defined as PANoptosis, an inflammatory PCD pathway regulated by the PANoptosome complex, whose characteristics cannot be explained by any of the three PCD pathways. During viral infection, PANoptosis can promote inflammatory response by inducing the production of inflammatory cytokines and cell death to exert an antiviral mechanism. This article reviews the various effects of cell death pathways during viral infection and provides new ideas for clinical antiviral therapy and related immunotherapy.
Introduction
New viral infections pose a major global health threat, and the COVID-19 pandemic has infected hundreds of millions of people. In recent years, there have been epidemics of influenza, Ebola, and Zika viruses on a global scale, raising concerns about the struggle between humans and the virus again and again. After the virus infects the human body, it induces the body to produce complex antiviral immunity. As the body's first line of defense against viral infections, the innate immune pathway plays an important role in the process of viral infections. Programmed cell death (PCD) is a key component of innate immunity. In recent years, the PCD pathways that have been clearly described include pyroptosis, apoptosis, and necroptosis, each of which can be activated during viral infection (36,60,72,85).
Pyroptosis is a form of inflammatory cell death characterized by lysis. It is mediated by inflammasomes, including typical inflammasomes activated by caspase-1 and atypical inflammatory bodies activated by caspase-4/5 or caspase-11 in mice. These inflammasomes act on a pore-forming protein gasdermin D (GSDMD), which drives cell leakage and lysis on the one hand and promotes the release of cytokines interleukin (IL)-1β and IL-18 on the other hand. Apoptosis was first considered as “immune silence” PCD, but recent studies have shown that it is also inflammatory. Apoptosis proceeds through a proteolytic signal cascade mediated by the caspase family (caspases) and occurs through two signal pathways: caspase-9-dependent endogenous pathway or caspase-8-dependent exogenous pathway. The consequences of apoptosis lead to cell disintegration, including chromosome condensation, DNA fragmentation, and membrane bubbles to form apoptotic bodies, which are finally swallowed and cleared. Necroptosis is triggered downstream of death domain receptors (such as tumor necrosis factor receptor [TNFR] and Fas) and Toll-like receptor (TLR)-4 or TLR3. It interacts with receptor interaction serine/threonine kinase 1 (RIPK1) and RIPK3 to activate downstream mixed lineage kinase domain like pseudokinase (MLKL) phosphorylation, thereby forming Necrosis, leading to cell necroptosis (8,34,73,84,87).
Initially, the virus invades the cell and causes the death of the host cell, which is considered harmful to the host. In fact, cell death caused by virus infection is a double-edged sword. In some tissues, it can increase cell destruction without loss of function. At this time, it serves the host and promotes virus clearance; virus-induced cell death is delayed, or selective killing of immune cells leads to immune deficiency, as shown in the example of HIV-infected CD4+ T cells, which are undoubtedly harmful to the host. As more and more studies have found that there are complex interrelationships among the three PCDs, a more detailed understanding of PANoptosis is essential for formulating new antiviral strategies to combat new viral infections and even possible virions that may appear in the future.
Pyroptosis
Definition of pyroptosis
Pyroptosis is a form of lytic and inflammatory cell death induced by caspase (79). The earliest research on pyroptosis can be traced back to 1986. Since then, Cerretti et al. and Thornberry et al. observed that ICE (IL-1β-converting enzyme, caspase-1) is an inflammatory caspase that can process Pro-IL-1β into mature IL-1β (9,76). Studies have found that in macrophages lacking the gene encoding caspase-1, cell death induced by Streptococcus freundii was eliminated (33). Until 1998, some scholars discovered the caspase-dependent cell death pathway in macrophages again (33). These findings together reveal the role of caspase-1 in bacterial-induced cell death. Since previous studies have shown that caspase-1 can mediate pro-IL-1β and pro-IL-18 protein cleavage of pro-inflammatory precursor cells (45), the term “pyroptosis” was subsequently proposed to define caspase-1-dependent inflammatory cell death (7,16,56). The definition of pyroptosis has now been expanded to include cell death mediated by most inflammasome caspases (including caspase-1 and caspase-4/5) (caspase-11 in mice).
Pyroptosis is mediated by an inflammasome, which is a multiprotein complex containing sensor protein, adaptor protein ASC (the adapter apoptosis-associated speck-like protein containing a caspase recruitment domain [CARD]), and caspase-1. A typical inflammasome sensor usually contains a caspase activation and recruitment domain (CARD) (e.g., NLRC4, NLRP1b) or pyrin domain (PYD) (e.g., NLRP3, AIM2, Pyrin) (53,56), which are essential for initiating the assembly of the inflammasome.
The molecular mechanism of pyroptosis
The classic pathway of pyroptosis
The classical pyroptosis pathway is mediated by inflammasomes, accompanied by the lysis of GSDMD and the release of IL-1β and IL-18. Inflammasomes are multiple complexed, most of which consist of proteins rich in leucine repeat sequences (NOD-like receptors, NLRs), ASC containing CARD, and pro-caspase-1. When viruses invade host cells, cytoplasmic pattern recognition receptors (PRRs, also called inflammasome sensors) recognize pathogen-related molecular patterns and danger-related molecular patterns (DAMPs). After activation, PRRs assemble into inflammasomes with pro-caspase-1 and ASC. A typical inflammasome sensor usually contains a CARD (e.g., NLRC4, NLRP1b) or PYD (e.g., NLRP3, AIM2, Pyrin) (91), which can be divided into NLRPs or NLRCs according to whether their N-terminal contains PYD or CARD. NLRPs contain one or more PYDs at their N-terminus, such as NLRP1 and NLRP3, while NLRCs contain CARD.
ASC contains a PYD and a CARD. CARD is necessary for the recruitment of pro-caspase-1 to form an inflammasome. ASC connects the inflammasome sensor containing PYD or CARD with the caspase-1 containing CARD through homotypic domain interaction. After the inflammasome is assembled, caspase-1 is activated to become mature caspase-1. On the one hand, caspase-1 cleaves the executive protein GSDMD to form N-GSDMD, which penetrates the cell membrane to form nonselective pores, leading to cell swelling and pyroptosis (11,67,70). On the other hand, caspase-1 cleaves pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, which are released through the pores formed by N-GSDMD, causing pyroptosis (30,76).
The activation of these IL-1 family cytokines is of great significance to the immune stimulation downstream of pyroptosis. IL-18 can stimulate CD4+T to produce interferon (IFN)-γ1 type helper T cells (Th1) and induce natural killer (NK) cell responses, while IL-1β can support the expansion of T cells and enhance the effect of T cells on B cells to stimulate antibody production and support the differentiation of Th17 cells.
Nonclassical pathways of pyroptosis
In the nonclassical pyroptosis pathway, human caspase-4/5 or mouse caspase-11 is activated by directly binding to intracellular lipopolysaccharide through the N-terminal CARD. Activated caspase-4/5/11 can also cleave GSDMD into N-GSDMD, which oligomerized and transferred to the cell membrane, eventually forming plasma membrane pores. Activated caspase-4/5/11 cannot cleave pro-IL-1β/pro-IL-18 but can promote the activation of NLRP3 inflammasome and caspase-1 in an endogenous manner, thereby inducing IL-1β and IL-18 secretion. In addition, GSDMD is cleaved by caspase-4/5/11, resulting in K + efflux, inducing the assembly of NLRP3 inflammasomes, and ultimately leading to pyroptosis.
Other pathways of pyroptosis
Recent studies have found that caspase-3/8 and granzyme B (GzmB) can also mediate pyroptosis. In the caspase-3 mediated pathway, active caspase-3 cleaves gasdermin e (GSDME) to form N-GSDME, which induces pyroptosis. In the caspase-8-mediated pathway, inhibition of TGF-β-activated kinase 1 induces the lysis of caspase-8-related GSDMD, leading to pyroptosis. Recently, studies have also reported the pyroptosis mediated by Gzm. It was found that chimeric antigen recepter T cells rapidly activate caspase-3 in target cells by releasing GzmB and then activate GSDME, which in turn causes pyroptosis. In addition, GzmA and GzmB in cytotoxic lymphocytes can also induce pyroptosis by entering target cells through perforin. These latest studies have broadened our understanding of the pyroptosis pathway; however, the mechanism of the new pathway still needs to be elaborated in more detail.
Pyroptosis during virus infection
Virus infection drives pyroptosis through the activation of inflammasome
Since numerous studies have found that pyroptosis plays an important role during virus infection, a large number of studies have explored the specific mechanism. Studies on multiple virus infection models have found that multiple virus infections can drive pyroptosis through the activation of inflammasomes.
Coronavirus infection: activation of inflammasomes has been found in coronavirus infection, including the newly emerged SARS-CoV-2 (64). After SARS-CoV-2 infects human monocytes, it induces the formation of NLRP3, the lysis of caspase-1, and the secretion of IL-1β and IL-18, which in turn induces pyroptosis of PBMC (1). Studies have found that the inflammatory components in the plasma of COVID-19 patients, such as caspase-1, IL-18, lactate dehydrogenase, and GSDMD, are positively correlated with the severity of the disease (64). In addition, NLRP3-positive cells can be found in the lung tissue of patients who died of COVID-19.
Zika virus infection: in neural progenitor cells, Zika virus induces the lysis of caspase-1 and GSDMD and the release of IL-1β and IL-18 (31). The process is accompanied by the upregulation of the expression of NLRP3, while the expression of NLRC4, AIM2, or Pyrin remains unchanged, which indicates that the activation of inflammasomes induced by Zika virus infection may depend on NLRP3.
HIV infection: studies have found that after HIV infects CD4+ T cells, pyroptosis is activated by caspase-1. Blocking caspase-1 with the caspase-1 inhibitor VX-765 can significantly inhibit caspase-1 lysis, IL-1β release, and CD4+ T cell death. Inhibition of caspase-3 does not protect CD4+ T cells from death (21).
Semliki Forest virus (SFV) infection: in human keratinocytes and epithelial cells, NLRP1 acts as a double-stranded RNA (dsRNA) sensor during SFV infection. NLRP1 directly interacts with dsRNA through its leucine-rich repeat domain to induce ATP hydrolysis and trigger NLRP1 oligomerization, which subsequently leads to inflammasome activation and pyroptosis (2) (Fig. 1).
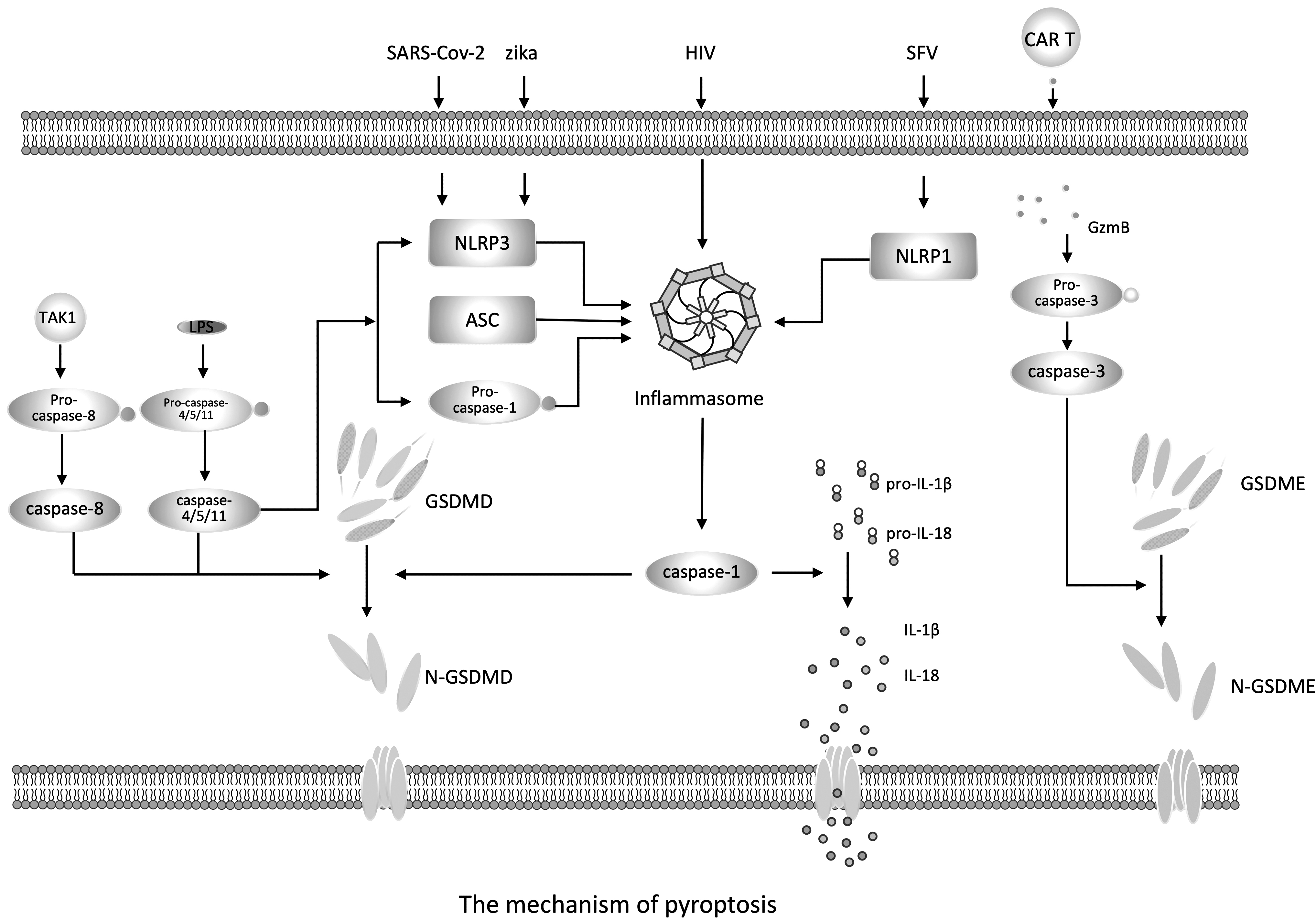
The mechanism of pyroptosis. Note: Viral invasion activates PRRs, which induce ASC and pro-caspase-1 to assemble into inflammasomes, forming the classic pathway of cell death. The typical inflammasome consists of proteins rich in leucine repeat sequences (NLRs), adaptor apoptosis-related dot-like protein (ASC), and pro-caspase-1. Upon assembly of the inflammasome, pro-caspase-1 is activated and become the mature caspase-1. On the one hand, caspase-1 cleaves the executive protein GSDMD to form N-GSDMD, which penetrates the cell membrane to form nonselective pores, resulting in cell swelling and pyroptosis. On the other hand, caspase-1 cleaves pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, which are released through the pores formed by N-GSDMD, causing pyroptosis. The nonclassical pathway of pyroptosis is mediated by caspase-4/5 (caspase-11 in mice). Activated caspase-4/5/11 can also cleave GSDMD into N-GSDMD, which eventually leads to pyroptosis. Activated caspase-4/5/11 can also promote the activation of NLRP3 inflammasome and caspase-1, thereby inducing IL-1β and IL-18 secretion. In addition, CAR T cells rapidly activate caspase-3 in target cells by releasing GzmB, which then activate GSDME, resulting in cell death. SARS-CoV-2 and Zika virus infection mainly induces pyroptosis by inducing the formation of NLRP3 and the cleavage of caspase-1, while HIV-infected CD4+ T cells activate pyroptosis through caspase-1. Blocking of caspase-1 with the caspase-1 inhibitor VX-765 significantly inhibits CD4+ T cell death. SFV induces inflammasome activation and pyroptosis by interacting with NLRP1. ASC, the adapter apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD); CAR, chimeric antigen receptor; GSDMD, gasdermin d; GSDME, gasdermin e; GzmB, granzyme-B; IL, interleukin; NLRs, NOD-like receptors; SFV, Semliki Forest virus.
Viruses directly interfere with pyroptosis
During viral infection, the inflammasome and the cytokines that the inflammasome depend on contribute to antiviral immunity and clearance of infection. As mentioned above, various DNA and RNA viruses induce pyroptosis through NLRP1 or NLRP3 inflammasomes during infection, and numerous studies have found overlap between pyroptosis and the release of IL-1β/IL-18, whereas the cytokine-independent role of pyroptosis in host defense has been poorly studied. Studies have proved that the production of IL-1 and IL-18 alone cannot fully explain the protective effect on the body (42). For example, the mortality rate of the IL1β-/-IL18-/- mice central nervous system infected with attenuated rabies virus strains is higher compared with caspase-1-/-caspase-11-/-mice, which means that the host protection mechanism requires caspase-1/-11 induced IL-1 family function.
Although there are many examples of inflammasomes inducing pyroptosis during viral infection, viruses can also directly interfere with pyroptosis by regulating inflammasome activation or targeting GSDMD.
In Sendai virus (SeV) infection, SeV V protein can directly bind to NLRP3 and inhibit NLRP3's own oligomerization, resulting in a decrease in IL-1β release (44). The V protein of human parainfluenza virus type 2 and Nipah virus can also interact with NLRP3 to inhibit inflammasome activation (44). In enterovirus 71 (EV71) infection, compared with wild-type mice, mouse lines with NLRP3 inflammasome activation deficiency, such as NLRP3-/-, ASC -/-, and caspase 1-/- mice, will have more severe viral infection (83), indicating that NLRP3 inflammasome activation is important for the host's defense against EV71 infection. To overcome this host defense strategy, EV71 protease 3C interacts with NLRP3 and cleaves NLRP3 to inhibit the activity of NLRP3 inflammasomes, thereby exacerbating the occurrence of viral infections (48). EV71 protease 3C also cleaves GSDMD to form an N-terminal fragment, which cannot trigger pyroptosis, indicating that EV71 protease 3C can prevent pyroptosis by inactivating GSDMD.
Apoptosis
Definition of apoptosis
Apoptosis is the first PCD described, which has been described morphologically as early as 1972 (40). Cells undergoing apoptosis exhibit morphological changes, including nuclear and chromatin condensation, DNA fragmentation, and membrane blistering to form apoptotic bodies, which are eliminated after being swallowed by phagocytes (28). It is now known that apoptosis proceeds through endogenous and exogenous pathways. The identification of apoptotic peptidase activator 1 (APAF1) confirmed the endogenous, mitochondrial-dependent pathway of apoptosis. Mitochondrial damage or destruction leads to the mitochondrial outer membrane permeability (MOMP), resulting in the release of many molecules, including cytochrome C. Cytochrome C is recognized by APAF1 and then forms apoptotic bodies with caspase-9, which is the initiation caspase of the endogenous apoptotic pathway (41). This complex allows the caspase-9 precursor to be cleaved into its mature form through a mechanism related to ATP hydrolysis. Then mature caspase-9 induces the activation of caspase-3 and -7 to drive apoptosis (49).
Soon after describing the endogenous apoptosis pathway, the exogenous pathway was also identified. Exogenous apoptosis is initiated by extracellular signals through the binding of death ligands such as Fas ligand (FasL), TNF, and TNF-related apoptosis-inducing ligand (TRAIL) to their respective receptors (12). The first pro-apoptotic signaling molecules downstream of these death receptors are Fas-associated death domain (FADD) and caspase-8. FADD interacts with the receptor DD and recruits caspase-8 through homotypic interactions between its death effect domains. Caspase-8 is recruited in its prototype form and processed automatically into mature caspase-8 to obtain its complete proteolytic activity. Active caspase-8 can cleave and activate caspase-3 and caspase-7 (5). Therefore, caspase-9 and caspase-8 are the initial caspases of the endogenous and exogenous apoptotic pathways, respectively, which converge to activate the same executive enzymes: caspase-3 and caspase-7.
Endogenous cell apoptosis
Endogenous apoptosis is activated by intracellular stress signals (such as DNA damage, etc.). Endogenous apoptosis is triggered when endogenous stress signals affect the balance of B-cell lymphoma-2 (Bcl-2) protein family (50). Endogenous apoptosis is induced by the balance of Bcl-2 protein family affected by the signal of internal stress (such as DNA damage, etc.) in the cell.
The Bcl-2 family includes pro-apoptotic members (BCL-2-associated X protein [BAX], BCL-2-antagonistic killer [BAK], BID, Bcl-2-interacting mediator of cell death [BIM], etc.) and antiapoptotic members (BCL-2, BCL-XL, MCL-1). Pro-apoptotic Bcl-2 can trigger MOMP (permeabilization of the outer mitochondrial membrane), which in turn induces the release of the second mitochondrial activator of caspases (SMAC) and the electron transport chain component cytochrome C (24). SMAC antagonizes X-linked inhibitor of apoptosis (XIAP), which inhibits apoptosis of caspases-9, -3, and -7. The released cytochrome C is bound by the cell membrane sensor APAF-1, which recruits and activates caspase-9, and the resulting multiprotein signaling complex is called apoptosome (49). Activated caspase-9 further induces the activation of caspase-3 and -7 to drive apoptosis (49).
Exogenous apoptosis
Contrary to endogenous apoptosis, exogenous apoptosis is usually driven by extracellular signals (81), which are combined with death receptors in the TNF superfamily to achieve signal transduction, including the combination of TNFR1/2 and TNF, Fas/CD95 and FasL, or TRAILR and TRAIL/Apo2 (80,82). Although these receptor-ligand pairs are different in composition, they can recruit a protein called FADD and then pro-caspase-8. Through homotypic interaction, pro-caspase-8 is activated. On the one hand, activated caspase-8 can directly activate caspase-7 and -3 to induce cell apoptosis. On the other hand, it can also indirectly induce endogenous apoptosis by cleaving the pure BCL-2 homology domain 3 (BH3) protein BH3 interacting domain death agonist (BID) into an active form of tBid and driving the apoptosis induced by BAK/BAX mediated MOMP (13,65).
Apoptosis during virus infection
Viruses are intracellular pathogens, which must rely on host cells to replicate and spread. Therefore, clearing virus-infected cells through any form of PCD is an effective host defense strategy, which not only controls infection but also exposes virus-related immune molecules. In addition, other signal molecules produced by specific PCD pathways also further promote the inflammatory response and antiviral response (37). Apoptosis pathway is the most studied PCD in virus infection. Due to the expression of many inflammation-inhibiting molecules during the process, apoptosis was once considered to have poor immunity (19), but it still plays an important role in eliminating virus-infected cells.
Cytotoxic immune cells induce apoptosis
Apoptosis is the main PCD pathway for cytotoxic immune cells to eliminate infected cells. These cells are essential for controlling the infection of intracellular pathogens such as viruses. These cells include cytotoxic T lymphocytes (CTL) and NK cells. CTL and NK cell-driven cell killing is dependent on the apoptotic pathway and is driven by the secretion of cytotoxic perforin and Gzm and the death receptor Fas (also known as CD95) (52). CTL and NK cells first recognize the virus-infected cells and target the release of cytotoxic particles. GzmB (serine protease B) is stored in this cytotoxic granule, and GzmB enters target cells through the formation of transmembrane pores or endocytosis (52). Subsequently, GzmB cleaves BH3 to cause BAX/BAK mediated MOMP and initiate caspase-9-driven endogenous apoptotic pathway (62). The combination of FasL expressed on the surface of CTL and the FAS receptor on the target cell leads to the recruitment of FADD, which in turn recruits caspase-8, and induces exogenous apoptosis of virus-infected cells (14).
Viral genome mediates cell apoptosis
In addition to cell death induced by cytotoxic immune cells, it has been found that the activation of PRRs can directly lead to the initiation of cell-autonomous apoptotic pathways. The viral genome can be sensed in many forms, including viral DNA (vDNA), viral single-stranded RNA, or dsRNA.
In the process of virus replication, intracellular dsRNA fragments can be produced, which can be recognized by two RNA helicases containing CARD on the cell membrane, namely: retinoic acid-induced gene 1 (RIG-1) and melanoma differentiation related gene-5 (MDA-5) (39). RIG-1 is activated after recognizing dsRNA and induces the activation of mitochondrial antiviral signal (MAVS) through CARD-CARD isotype interaction. MAVS can lead to the activation of IRF-3 and nuclear factor kappa B (NF-κB) in the next step (69). In the pathway of IRF-3, MAVS interacts with TNFR-related silver-3 (TRAF-3) to recruit TRAF-related NF-κB activators, which subsequently leads to phosphorylation of IRF-3 and -7 (3). This IRF-3 signaling pathway can produce IFN-I and IFN-III, then activate CTL and NK cells, and then induce cell apoptosis. This signal transduction has a positive regulatory effect on cell apoptosis.
The extracellular vRNA can be recognized by the cell membrane TLR3. TLR3 recruits IKK (IκB kinase) through the adaptor protein Toll/TIR (IL-1 receptor), which induces the activation of IRF-3. In contrast, TLR3 can also mediate the activation of NF-κB through RIPK1. In turn, it induces the production of type I IFNs and cytokines and promotes apoptosis (26,57).
Since DNA is only present in the nucleus under normal circumstances, the vDNA in the cytoplasm will also trigger the induction signal. Cyclic GMP-AMP synthase (cGAS) can bind to vDNA to produce circulating GMP-AMP (cGAMP), leading to the activation of stimulator of interferon genes (STING), which in turn induces IRF-3 activation and apoptosis (20,35,74) (Fig. 2).
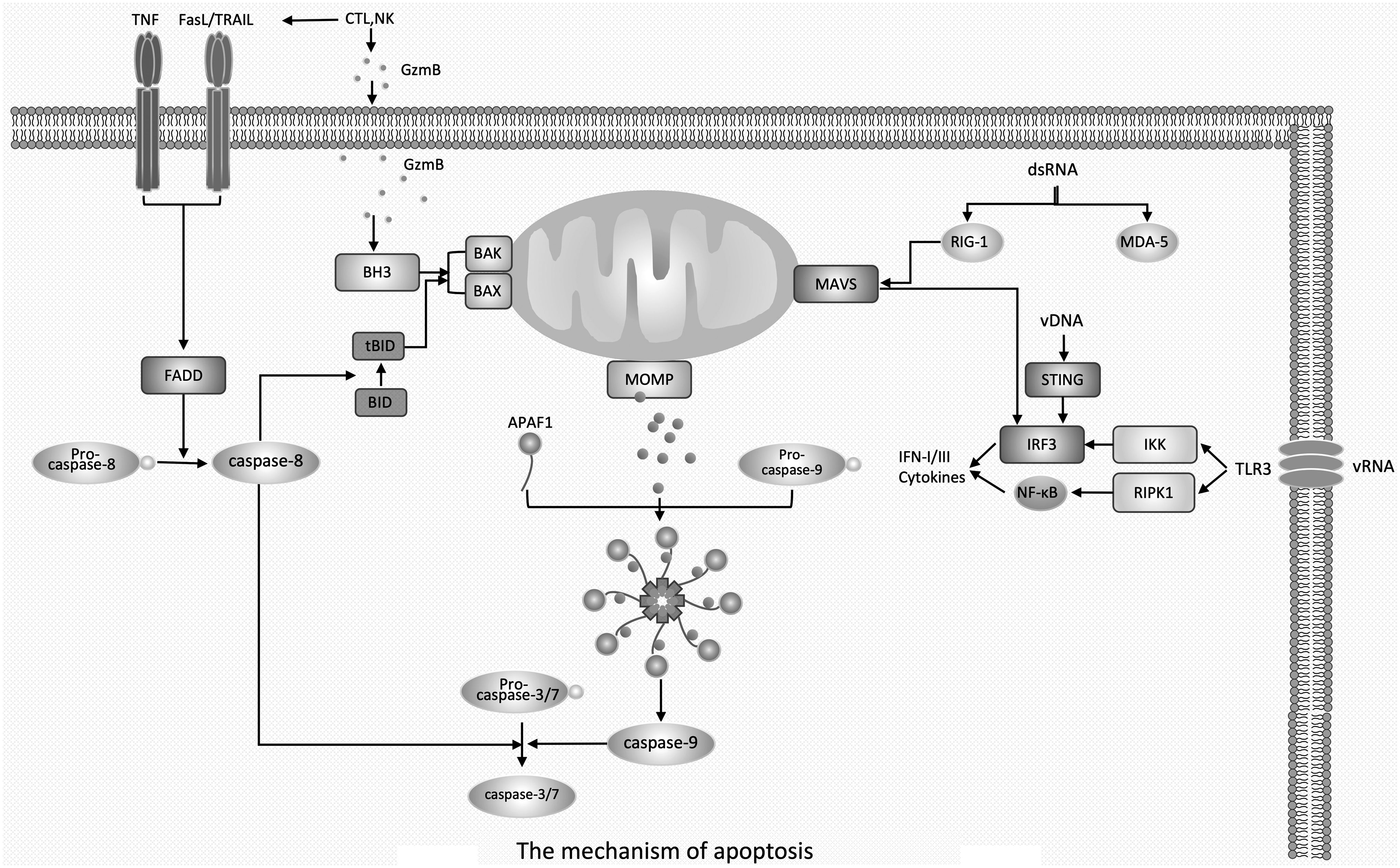
The mechanism of apoptosis. Note: Intracellular stress signals trigger the pro-apoptotic members BAK/BAX of the Bcl-2 family, which induces MOMP. Cytochrome C in mitochondria is released and then bound by the cell membrane sensor APAF-1, which in turn recruits and activates caspase-9. Activated caspase-9 further induces the activation of caspase-3 and -7, leading to membrane blebbing and apoptosis. In contrast to endogenous apoptosis, exogenous apoptosis is usually driven by extracellular signals. Exogenous signals bind to death receptors in the TNF superfamily of receptors, recruit FADD proteins, and subsequently induce activation of pro-caspase-8. Activated caspase-8, on one hand, directly activates caspase-7 and -3 to induce apoptosis, and on the other hand, it also cleaves BID to its active form tBID and drives BAK/BAX-mediated MOMP-induced apoptosis, thereby indirectly inducing endogenous apoptosis. CTL and NK release GzmB into target cells to initiate endogenous apoptosis through BH3-induced BAK/BAX-mediated MOMP response. In addition, CTL express FasL that bind to FAS receptors on target cells, leading to the recruitment of FADD, which in turn activates caspase-8 and induces exogenous apoptosis. dsRNA is recognized by RIG-1 and MDA-5 on the cell membrane and induces the activation of IRF-3 through MAVS, which promotes the production of IFN-I and IFN-III, and then activates CTL and NK cells, thereby inducing apoptosis. In addition, extracellular vRNA can be recognized by membrane TLR3, which activates IRF-3 and NF-κB using IKK and RIPK1, respectively. vDNA induces activation of IRF-3 using STING. All three of the above typical viral genome-induced apoptosis culminate in the activation of IRF-3, suggesting that IRF-3 plays a critical role in inducing apoptosis in viral infection. APAF1, apoptotic peptidase activator 1; BAK, BCL-2-antagonistic killer; BAX, BCL-2-associated X protein; Bcl-2 family, B-cell lymphoma-2 protein family; BID, BH3 interacting domain death agonist; BH3, BCL-2 homology domain 3; CTL, cytotoxic T lymphocytes; dsRNA, double-stranded RNA; FADD, Fas-associated death domain; FasL, FAS ligand; IFN, interferon; IKK, IκB kinase; MAVS, mitochondrial antiviral signal; MDA-5, melanoma differentiation related gene-5; MOMP, mitochondrial outer membrane permeability; NK, natural killer; RIG-1, retinoic acid-induced gene 1; RIPK1, receptor interaction serine/threonine kinase 1; STING, stimulator of interferon genes; tBID, truncated BH3 interacting domain death agonist; TNF, tumor necrosis factor; vDNA, viral DNA.
The cell apoptosis induced by the above three typical viral genomes all take the activation of IRF-3 as the final result, which indicates that IRF-3 plays a key role in inducing apoptosis in viral infection.
The escape of virus in apoptosis
In the process of long-term game with viruses, the host has evolved extraordinary antivirus capabilities. Virus invasion will trigger a series of complex reactions in the host to eliminate it. However, in the struggle with the host, to ensure their own survival, viruses have evolved complex immune escape mechanisms. Many viruses encode virulence proteins that inhibit cell apoptosis, such as caspase inhibitors. Studies have found that the murine cytomegalovirus (MCMV) vICA protein can inhibit caspase-8. The possible mechanism is that the M36 protein encoded by MCMV blocks the exogenous apoptosis pathway induced by caspase-8 (23,71). The vaccinia virus CrmA protein can escape host cell death by inhibiting caspase-8 and GzmB (90). Herpes simplex virus (HSV) ribonucleic acid reductase ICP10 inhibits cell apoptosis by directly interacting with caspase-8 (22). In addition to caspase inhibitors, viruses can also encode BCL-2 homologs to block BAK and BAX mediated MOMP-dependent apoptosis, such as adenovirus E1B19K and Epstein–Barr virus BHRF1 and BALF1 (32,47).
Although the virus tries to evade cell death, the virus' blocking of apoptosis usually causes the host cell to activate other PCD pathways, indicating the interconnection between different pathways of PCD, which will be discussed in detail later in this review.
Necroptosis
Definition of necroptosis
In addition to pyroptosis and apoptosis, necroptosis has also been defined by molecules. Evidence of necroptosis was first reported in 2000 (34). In the presence of caspase inhibitors, T cells stimulated with FasL die in a FADD/RIPK1-dependent manner, which have a necrotic morphology, and do not release cytochrome C (34). In TNF-α-induced necroptosis, the kinase activities of RIPK1 and RIPK3 are critical for cell death; RIPK1 and RIPK3 interact through their RIP homotypic interaction motif (RHIM) to form necrosomes (16). Necrosomes phosphorylate MLKL (Pseudo Kinase Mixed Kinase Domain pseudo-kinase mixed lineage kinase domain-like), thereby destroying the plasma membrane and causing cell death (87). In addition, TLR can also initiate necroptosis through phosphorylation of RIPK3 and MLKL, regardless of RIPK1 kinase activity (89). Since the occurrence of necroptosis is usually related to the inhibition of caspases (especially caspase-8), necroptosis is considered to be a “fail-safe” mechanism to ensure the continuation of cell death under the condition that apoptosis is inhibited during infection (58).
The mechanism of necroptosis
The classic pathway of necroptosis
Necroptosis is morphologically manifested as cytoplasmic swelling and early membrane rupture and promotes the extracellular release of DAMP. Since the initiation of necroptosis occurs when TNFR is connected, and NF-κB is activated through the assembly of the ubiquitin framework complex (Complex1), the classic necroptotic pathway is called TNFR-dependent necroptosis pathway (86). Complex1 consists of TRAF2 and TRADD (TNF receptor type 1 related death domain) aptamers, IAPs (E3 ubiquitin ligase) and LUBAC (linear ubiquitin chain assembly complex), and RIPK1, among which RIPK1 plays a decisive role (18).
When caspase-8 is inhibited, RIPK1 can interact with RIPK3 through its RHIM to form a necrosome complex. The assembly of necrosome complexes leads to the phosphorylation of MLKL, which drives MLKL to undergo oligomerization, and then forms pores on the whole cytoplasmic membrane, resulting in osmotic pressure imbalance and membrane rupture, that is, cell necroptosis (73). RIPK1 and RIPK3 play a central role in the regulation of necroptotic pathways, and activated caspase-8 can cleave them. Therefore, the inhibition of caspase-8 is an absolute prerequisite for initiating the RIPK1-RIPK3 necrosome complex (75).
A new approach to necroptosis
Similar to RIPK, the nucleic acid sensing receptor ZBP1 has a RHIM, so ZBP1 can interact with RIPK3 (78). Then RIPK3 can initiate the phosphorylation of MLKL and induce necroptosis. Therefore, this pathway can be called the ZBP1-dependent necroptosis pathway. As mentioned earlier, because RIPK3 can induce MLKL-dependent cell apoptosis, ZBP1 can initiate both the apoptotic pathway and the necroptotic pathway at the same time. As a form of immunogenic cell death, necroptosis can stimulate the host to produce a strong immune response, while apoptosis is considered to be very weakly immunogenic. Therefore, during viral infection, the special effect of ZBP1 can be used to fine-tune the cell death pathway to achieve a new balance, which can not only enhance the host's antiviral immunity, but not cause excessive immunity to damage the host (46). Of course, this requires more in-depth research on the new pathway of necroptosis.
Necroptosis during virus infection
After the death receptor TNFR is activated by the TNF ligand, it forms the ubiquitin framework complex (Complex1) which activates NF-κB to express survival and inflammatory signals. This survival signal is mediated by deubiquitinase A20 and/or cylindromatosis (a RIPK1 K63 deubiquitinating enzyme), triggering the formation of different cell death complexes, such as TRADD-FADD-caspase-8 or TRADD-RIPK1-FADD-caspase-8, to induce cell apoptosis (86).
As mentioned above, RIPK1 can interact with RIPK3 through its RHIM to form a necrosome complex during viral infection or when caspase-8 is inhibited. Necrosome initiates RIPK3 to mediate MLKL phosphorylation and change its conformation. Phosphorylated MLKL oligomerizes into a polymer complex, which in turn destroys the plasma membrane and leads to cell necroptosis. Under most physiological conditions, the formation of the RIPK1-RIPK3 necrosome complex is inhibited by the cFLIP/caspase-8 heterodimer complex, which is recruited into the RIPK1-RIPK3 complex through FADD and cleaves RIPK1 and RIPK3 to promote apoptosis (61). Therefore, the inhibition of caspase-8, such as the response to viral effectors (such as the MCMV vICA protein), is a key event that triggers necroptosis (71). Similar to the apoptosis pathway, intracellular viral nucleic acids can be recognized by cGAS during viral infection. IFNs are induced by the cGAS-STING pathway and thus promote necroptosis (4,6,10,68).
In addition, it has been proven that during viral infection, necroptosis can also be activated using a ZBP1-dependent pathway, leading to RIPK3-induced MLKL phosphorylation, which in turn causes cell death (78). It has been reported that ZBP1-mediated cell death occurs in many viral infections (14,17,78) (Fig. 3). In addition to its role in necroptosis, ZBP1 is also involved in the entire PCD pathway, which will be discussed in depth in the next section.
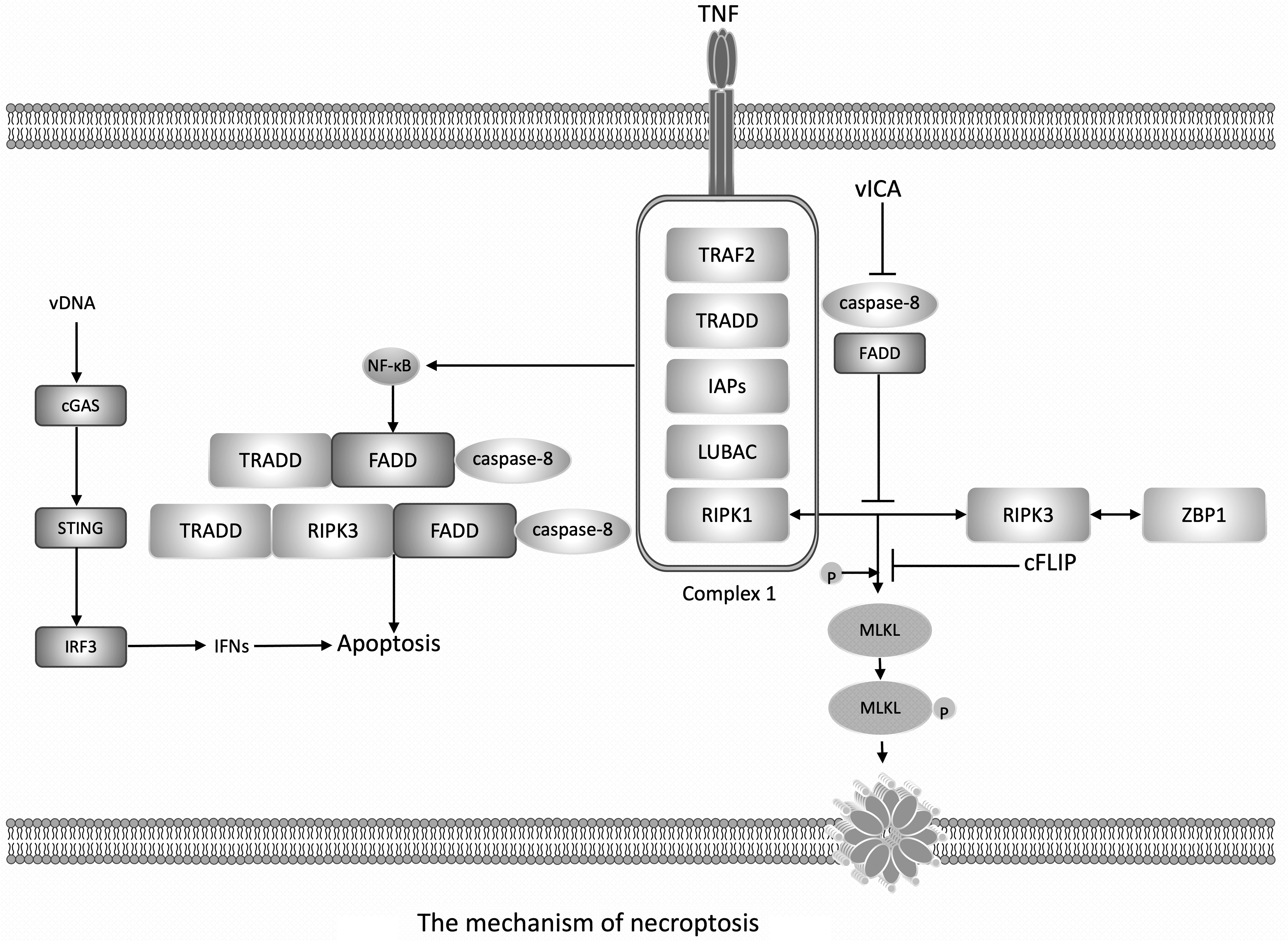
The mechanism of necroptosis. Note: The initiation of necroptosis occurs when TNFR is connected, and NF-κB is activated through the assembly of the ubiquitin framework complex (Complex1). Complex1 consists of TRAF2 and TRADD aptamers, IAPs (E3 ubiquitin ligase) and LUBAC, and RIPK1. When caspase-8 is inhibited, RIPK1 can interact with RIPK3, then leads to the phosphorylation of MLKL. Phosphorylated MLKL undergoes oligomerization and then forms pores on the whole cytoplasmic membrane, resulting in osmotic pressure imbalance and membrane rupture, that is, necroptosis. RIPK1 and RIPK3 play a central role in the regulation of necroptotic pathways, and activated caspase-8 can cleave them. Therefore, the inhibition of caspase-8 is an absolute prerequisite for initiating the RIPK1-RIPK3 necrosome complex. ZBP1 has a RHIM, so ZBP1 can interact with RIPK3, then inducing necroptosis. During viral infection, the death receptor TNFR is activated by the TNF ligand, it forms the ubiquitin framework complex (Complex1) which activates NF-κB, then NF-κB triggering the formation of different cell death complexes, such as TRADD-FADD-caspase-8 or TRADD-RIPK1-FADD-caspase-8, to induce cell apoptosis. As mentioned above, RIPK1 can interact with RIPK3 through its RHIM to form a necrosome complex during viral infection or when caspase-8 is inhibited. During viral infection, intracellular viral nucleic acids can be recognized and promote necroptosis through the cGAS-STING pathway. In addition, during viral infection, necroptosis can also be activated through a ZBP1-dependent pathway. cGAS, cyclic GMP-AMP synthase; IAP, inhibitor of apoptosis; LUBAC, linear ubiquitin chain assembly complex; NF-κB, nuclear factor kappa B; MLKL, mixed lineage kinase domain like pseudokinase; RHIM, RIP homotype interaction motif; TNFR, TNF receptor; TRADD, TNF receptor type 1 related death domain.
The escape of viruses in necroptosis
Like the apoptosis pathway, many viruses have evolved viral proteins to inhibit cell necrosis and induce immune escape. Vaccinia virus viral protein E3L has an N-terminal Zα domain similar to ZBP1, which can compete with ZBP1 to inhibit ZBP1-RIPK3 and prevent necroptosis (43). In addition, the MCMV viral protein M45 contains a RHIM domain, which can interfere with the interaction between RHIM-ZBP1 and RIPK3, leading to the inhibition of necroptosis (78). Recent evidence also shows that some viruses can lead to the elimination of RIPK3, such as the virus inducer (vIRD), which can degrade the RIPK3 of cowpox virus and induce the protein degradation of RIPK3 to inhibit necroptosis (51).
Interaction Between PCD During Virus Infection
Definition of PANoptosis
Initially, the three pathways of PCD were considered to be different and independent of each other, but with the continuous deepening of research, more and more evidence shows that there are extensive interactions among these PCD pathways (60,63,85). These findings led to the establishment of the concept of PANoptosis, which is defined as a type of inflammatory PCD activated by a specific trigger and regulated by the PANoptosome complex. It has the key characteristics of pyroptosis, apoptosis, and/or necroptosis, but it cannot be explained by any of these PCD pathways (60,63,85) (Fig. 4). Studies have shown that pyroptosis cleaves the caspase and ADP-ribose polymerase 1 (PARP1) in the apoptotic pathway through caspase-1. The apoptotic caspase-8 is very important for the activation of NLRP3-dependent inflammasomes. As mentioned, both cell apoptosis and necroptosis are regulated by the activity of caspase-8 (27). In fact, PANoptosome is equivalent to providing a molecular scaffold that allows the interaction and activation of the key molecules of pyroptosis (ASC, caspase-1), apoptosis (caspase-8), and necroptosis (RIPK1, RIPK3).

The mechanism of PANoptosis. Note:
Under physiological conditions, classic signals from death receptors can trigger exogenous cell apoptosis by activating caspase-8, which in turn also cleaves RIPK1 or/and RIPK3 to prevent necroptosis. Caspase-8 is also involved in mediating the initiation and activation of classic and nonclassical NLRP3 inflammasomes (29). Caspase-8 can bind to ASC and induce lysis of GSDMD to trigger pyroptosis (55). In the absence of caspase-1, ASC-dependent activation of caspase-8 can also trigger cell apoptosis (66). NLRP3 and NLRC4 inflammasomes can cause the cleavage of the apoptosis marker PARP1 (54). In addition, in the absence of GSDMD, inflammasome-induced caspase-1 activation can trigger cell apoptosis through BID, caspase-9, and caspase-3. Activated caspase-3 can also cleave GSDME and cause pyroptosis (77). The formation of PANoptosome complex further promotes the activation of downstream cell death effectors, including pyroptosis (caspase-1 and GSDMD), apoptosis (caspase-3/7), and necroptosis (RIPK3 and MLKL).
Therefore, in some virus infection models, all key components of PANoptosome need to be deleted to protect host cells and reduce cell death, which indicates that PANoptosome plays an important role in controlling virus infection.
PANoptosis during influenza A virus infection
During viral infection, especially during influenza A virus (IAV) infection, the evidence for the interaction between PCD pathways is more sufficient (46). Therefore, many studies have used IAV infection as a model of PANoptosis in host defense.
During IAV infection, ZBP1 interacts with RIPK3 through the RHIM domain and recruits caspase-8 and NLRP3 to promote the assembly of the PANoptosome complex, then driving PANoptosis. The mechanism is that ZBP1, as the vRNP sensor of IAV, induces the activation of NLRP3 and capsase-1-dependent inflammasomes, which in turn leads to the maturation of the cytokines IL-1β and IL-18, that is, the pyroptosis pathway. In addition, IAV infection can also cause the activation of caspase-8, caspase-3, and caspase-7. As mentioned earlier, these caspases can induce apoptosis. Using pan-caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone to inhibit caspase activity during IAV infection can promote necroptosis.
Further studies have found that the loss of ZBP1 can inhibit the activation of all these PCD pathways, including NLRP3 inflammasome-induced pyroptosis, caspase-8-driven apoptosis, and RIPK1-RIPK3-induced necroptosis (46). However, the loss of any of the three PCD pathways alone does not prevent cell death (88). This indicates that ZBP1 is involved in pyroptosis, apoptosis, and necroptosis at the same time and is likely to be the main regulator of the inflammatory PCD pathway.
The role of PANoptosis in other viral infections
Although ZBP1 is a key factor involved in regulating PANoptosis during IAV infection, ZBP1 dependent PANoptosis does not seem to be activated in other RNA virus infections, including vesicular stomatitis virus (VSV), SeV, and respiratory syncytial virus (46). However, studies still have found that PANoptosis has been observed during many viral infections.
In VSV infection, deletion of caspase-1/11, GSDMD, GSDMD/MLKL, or RIPK3 cannot prevent cell death during VSV infection. In the case of combined deletion of caspase-8 and RIPK3, VSV-induced cell death is significantly reduced (15). It shows that the deletion of a single PCD pathway cannot prevent cell death, while the combined deletion of key components of PANoptosis can. The same situation can be seen in the following model. During mouse hepatitis virus (MHV) infection, caspase-8/RIPK3 is needed to induce PANoptosis, that is, blocking the pyroptosis pathway only by deleting NLRP3, caspase-1/-11, or GSDMD will not lead to increased cell death, while combined deletion of caspase-8/RIPK3 can inhibit cell death induced by MHV infection (89).
In addition, an example that deserves special attention is SARS-CoV-2, which drives PANoptosis by inducing signal transduction and cytokine release (38).
A significant feature of severe patients with COVID-19 infection is cytokine storm, which can cause severe inflammation and even death in these patients. The two key cytokines produced during the cytokine storm are TNF-α and IFN-γ (25). Increased circulating levels of TNF-α and IFN-γ are associated with poor patient prognosis. These two cytokines synergistically induce PANoptosis, which is characterized by activated pyroptosis (GSDME), apoptosis (caspase-8/3/7), and necroptosis (pMLKL), leading to tissue and organ damage (38). By injecting TNF-α and IFN-γ into mice, the clinical symptoms of COVID-19 can be simulated. Treatment of mice with specific neutralizing antibodies of TNF-α and IFN-γ can provide protective effect during SARS-CoV-2 infection, suggesting that these two cytokines and their induction of PANoptosis are drivers of COVID-19 pathology. The mechanism is probably mediated by a new type of PANoptosis signaling molecule, namely NO, which provides a new direction for us to further understand PANoptosis (38).
In short, the new discovery of the interconnection between the PCD pathways of PANoptosis provides a new idea for understanding the complexity of PCD during virus infection. The presence of the central link PANoptosome allows multiple cell death effectors to participate at the same time, which may reduce the virus's inhibition of cell death, favoring virus-induced cell death, thereby providing protection for the host. However, recent studies have also found that some viruses can encode proteins that interfere with PANoptosis signals. MCMV-encoded RIP-activated virus inhibitor is a product of the M45 gene, which is a potent cell death inhibitor containing RHIM, and can target to inhibit ZBP1, RIPK3, and RIPK1, thereby restricting the formation of PANoptosome to avoid PANoptosis. HSV1 encodes the RHIM-containing protein ICP6, which inhibits the ZBP1-dependent cell death pathway through the RHIM domain in human cells. These studies have shown that viruses can evade the PANoptosis effect of host cells. Therefore, blocking the virus escape mechanism may be one of the effective methods to treat viral infections in the future.
Summary
In the millions of years of human survival and evolution, there is always a game with pathogens. As an intracellular pathogen, viruses must rely on host cells to survive. Due to this special way of survival, the host must effectively eliminate the infected cells to curb the spread of the virus. As an important part of the host's innate immune response, PCD is an effective host defense strategy. The interconnection between different pathways of PCD has led to the emergence of PANoptosis, which is defined as an inflammatory PCD pathway regulated by the PANoptosome complex, which has the key characteristics of pyroptosis, apoptosis, and/or necroptosis, and cannot be explained by any of these three PCD pathways alone. The emergence of PANoptosis provides new ideas to prevent viruses from escaping host immunity. We must now not only consider the influence of typical cell death, pyroptosis, apoptosis, or necroptosis in the body but also the influence of mixed cell death methods.
In the process of clinical diagnosis and treatment or drug development, we hope to obtain stronger host protection and, thus, may need to induce stronger cell death. As mentioned above, in a specific immune environment, viruses can enhance another PCD pathway by inhibiting one PCD pathway. Using this kind of balance-restricted relationship between PCDs, can we fine-tune a target in PANoptosis to enhance cell death? Or add an additional cell death inducer to activate the part of PCD we want? Of course, the activation of the immune system is very strictly controlled, because excessive activation will cause systemic inflammation and tissue damage, which is harmful to the host, and systemic excessive inflammation is common in various infectious diseases (59). Therefore, it is very important to find an immune balance point that can not only retain the antiviral function but also reduce the excessive inflammatory response.
In addition, as mentioned earlier, it has been discovered that there are complex virus escape mechanisms in both the apoptosis and necroptosis pathways. In fact, many discoveries about the function of cell death are based on experiments with pathogens, many of which have been genetically modified to remove their normal host avoidance strategies. Therefore, to understand the relationship between the virus and PANoptosis in the human body, we need to consider the existence of immune escape. This is also very common in clinical research. We often encounter patients with unsatisfactory treatment results, which is likely to be the result of virus escape.
Finally, due to the large variety of viruses and their respective characteristics, many of the discoveries mentioned above are based on specific classic viral pathogens. Therefore, there is still a long way to go to fully explore the complex mechanism of PANoptosis during viral infection. However, in view of the important role and broad application prospects of PANoptosis in antiviral immunity, it is necessary and worthwhile to invest more energy to study it.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Beijing Municipal Science and Technology Commission (no. Z151100004015122), Beijing Science and Technology Commission (no. D161100002716002), Special Public Health Project for Health Development in Capital (2021-1G-4061 and 2022-1-2172), National Science and Technology Major Project of China (nos. 2017ZX10201201-001-006, 2017ZX10201201-002-006, and 2018ZX10715-005-003-005), Beijing Hospitals Authority Clinical medicine Development of Special Funding Support (nos. XMLX 201706 and XMLX 202127), the Digestive Medical Coordinated Development Center of Beijing Hospitals Authority (nos. XXZ0302 and XXT28).