
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a global pandemic. There are four structural proteins of the virus: spike, envelope, membrane, and nucleocapsid proteins. Various vaccines were designed and are effectively being used against the spike protein of the virus. However, several vaccine-related complications have been reported worldwide. Assuming that the structural integrity of the whole protein might be contributing to these complications, this study was performed to design epitopes using the S2 domain of the spike protein, which could trigger a strong immune response. We have also predicted antigenic and allergenic properties of the selected epitopes. A total of 49 B cell epitopes passing antigenicity and other assessment filters were found using three methods. Among them, RDLICAQ had the highest antigenicity score (1.1443). However, only one cytotoxic T lymphocyte epitope, RSFIEDLLF, passed the essential filters with an antigenicity score of 0.5782 to show an appropriate immune response for T cells, while among 21 helper T cell lymphocyte epitopes that were filtered, FAMQMAYRFNGIGVT showed the highest (1.3688) antigenicity score. Conservation analysis revealed that the S2 domain is significantly conserved, thus making it an ideal candidate for vaccine development. We have also designed a vaccine construct based on the best suiting components found during the whole study. This construct and S2 domain solely can be future subjects of interest or might be included in a subunit cocktail formulation for attaining unabridged immunogenicity.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a global pandemic starting from Wuhan, China, in 2020. It is the third major human coronavirus from the Coronaviridae family after MERS-CoV and SARS-CoV. These viruses have high transmissibility and infectivity rates, which can cause a deadly infection. It was earlier thought that the virus originated from a fish market in Wuhan. It is enveloped and has a single-stranded RNA genome with a length of 29,881 nucleotide base pairs.
The genome codes for 9,860 amino acids. Four structural proteins of SARS-CoV-2 include spike, envelope, membrane, and nucleocapsid proteins, whereas there are 16 nonstructural proteins (NSPs), such as RNA-dependent RNA polymerase and papain-like protease (Huang et al, 2020).
The spike protein is 1,273 amino acids long and is present on the outer surface of the virus. It is mainly involved in the receptor recognition process and binding of the virus to the host cell. The angiotensin-converting enzyme (ACE2) receptor interacts with the spike protein to facilitate virus entry. S1 and S2 domains of the S protein are involved in receptor binding and membrane fusion, respectively (Wu et al, 2020).
Since a virus can neutralize antibodies by replacing just a single amino acid. Thus, for drug development, it is important to target those regions that are highly conserved; S2 being one such region, this study was designed to predict epitopes that have the ability to generate an immune response against it. We have also predicted antigenic and allergenic properties of selected epitopes. A conservation analysis in the S2 domain among different variants of SARS-CoV-2 was also performed. These variants included Alpha, Beta, Delta, Delta plus, Gamma, omicron, Omicron subvariant, Mu variant, and wild-type strain of SARS-CoV-2.
According to a study, two globally used vaccines, developed by Pfizer and AstraZeneca, were not so effective against the beta variant. The E484K mutation, found in the South African (B.1.351) variant and Brazilian (B.1.128) variant of SARS-CoV-2, was found to have a negative impact on neutralizing antibodies and thus decreased vaccine effectiveness (Madhi et al, 2021).
This shows that mutations, which can arise in future, can result in resistance to the virus and neutralize antibodies that are produced in response to currently available vaccines. Thus, it is important to recognize other viable options for future vaccine development.
Methods
Sequence retrieval and structural analysis of the S2 domain of the spike protein of SARS-CoV-2
The sequence of the S2 domain was retrieved from NCBI (Accession ID: QLI51913.1). PSIPRED (science, 2019) was used to predict the secondary structure of the S2 domain of the SARS-CoV-2 spike protein. The 3D structure of the S2 domain was predicted using PyMol and PDB ID 7FGCA. Physicochemical properties of the S2 domain such as half-life, molecular weight, and amino acid and atomic composition were analyzed using ProtParam (Bioinformatics, 2020).
The transmembrane topology of the S2 domain of the spike protein was checked through an online server, THMM (Denmark, 2022). The presence of disulfide bonds in the S2 domain was checked through an online tool, DiANNA (Clote, 2005).
Prediction of B cell epitopes
The B cell epitope prediction domain of IEDB (database, 2005) was used to predict B cell epitopes of the S2 domain of the SARS-CoV-2 spike protein. B cell epitopes were predicted using three methods: the BepiPred linear epitope prediction method, Emini surface accessibility method, and Kolaskar and Tongaonkar method. For checking whether the predicted epitopes are antigenic or not, VaxiJen, v2.0, was used (Flower, 2007).
Prediction of cytotoxic T cell lymphocyte epitopes
Cytotoxic T cell lymphocyte (CTL) epitopes were predicted using an online free access server, NetCTL 1.2 (Larsen, 2007), by setting the threshold value of 0.75. VaxiJen, v2.0, was again used to check the antigenicity of predicted epitopes. The ToxinPred server (Gupta, 2015) was used to check the toxicity of epitopes. The threshold value was set to 0.0. The Class I Immunogenicity tool of IEDB (Calis, 2013) was used to further check the immunogenicity of epitopes.
Prediction of helper T cell epitopes
The IEDB MHC II binding tool (Wang et al, 2010) was utilized for predicting helper T cells of the S2 domain of the SARS-CoV-2 spike protein. A 7-allele reference set was selected for prediction, which included seven Major Histocompatibility Complex, Class II, DR Beta alleles. The allergenicity of epitopes was checked using VaxiJen 2.0 (Flower, 2007) and toxicity was predicted using the ToxinPred server (Gupta, 2015).
MHC class I epitope prediction
ProPred-I (Singh, 2003) was employed for hunting MHC class I epitopes with a threshold value of 5%. Proteasome and immunoproteasome filters were switched “ON” with the threshold value of 5%. Then, the antigenicity of resultant epitopes was checked.
Prediction of MHC class II epitopes
ProPred (Singh, 2001) was assigned to predict epitopes of MHC class II. The threshold value was set to 4%. All of the alleles were selected for prediction. The antigenicity of predicted epitopes was determined.
Eminent features of the selected MHC class I and class II epitopes
Different properties such as digestion, mutation, toxicity, hydrophobicity, hydrophilicity, charge, pI, and mass of the resultant MHC class I and II epitopes, which passed the antigenicity test, were checked. An online tool, Protein Digest (biology), was used to check nondigesting enzymes of epitopes, while ToxinPred was utilized for checking toxicity (Gupta, 2015).
Conservation analysis between different countries
Sequences of eight different variants of the spike protein, region 686–1273, were downloaded from NCBI. Their accession numbers are as follows: wild-type strain—China (YP_009724390); Alpha variant—Austria (UFA39488); Omicron variant—USA: Arizona (UNN69607); Omicron subvariant—Bahrain (UNF17685); Delta—India (UMP57322); Beta—South Africa (ULL32733); Gamma—Paraguay (UNJ22161); and Mu variant—USA: Pennsylvania (QV034968).
Alignment of sequences was performed using Jalview 2.1.1.0 software. MEGA 7.0 was used to establish the phylogenetic tree.
Determination of conservation of selected epitopes
Conservation of all the selected epitopes that passed the assessment (i.e., antigenicity, immunogenicity, and toxicity) was checked using the epitope conservancy analysis tool of IEDB (Bui et al, 2007).
Multisubunit epitope vaccine design
For construction of the final vaccine construct, different filtered and final CTL, helper T cell lymphocyte (HTL), and B cell epitopes were used. Three separate linkers were used to join epitopes of three different types. CTL epitopes were joined with the help of the AAY linker, while HTL and B cell epitopes were linked together with GPGPG and KK linkers, respectively. Antigenicity of the vaccine was checked.
Results
Structural analysis of the S2 domain
The secondary structure of the S2 domain revealed that it consists mainly of a helix. After the helix, a major proportion consists of a coil and then a Beta strand, as shown in Figure 1A and B. 3D structure and conformation are given in Figure 2. Physiochemical structure analysis using ProtParam is given in Table 1. There were a total of 588 amino acids with grand average of hydropathicity (GRAVY) of 0.03.

Diagrammatic representation of the secondary structure of the S2 domain of the SARS-CoV-2 spike protein.
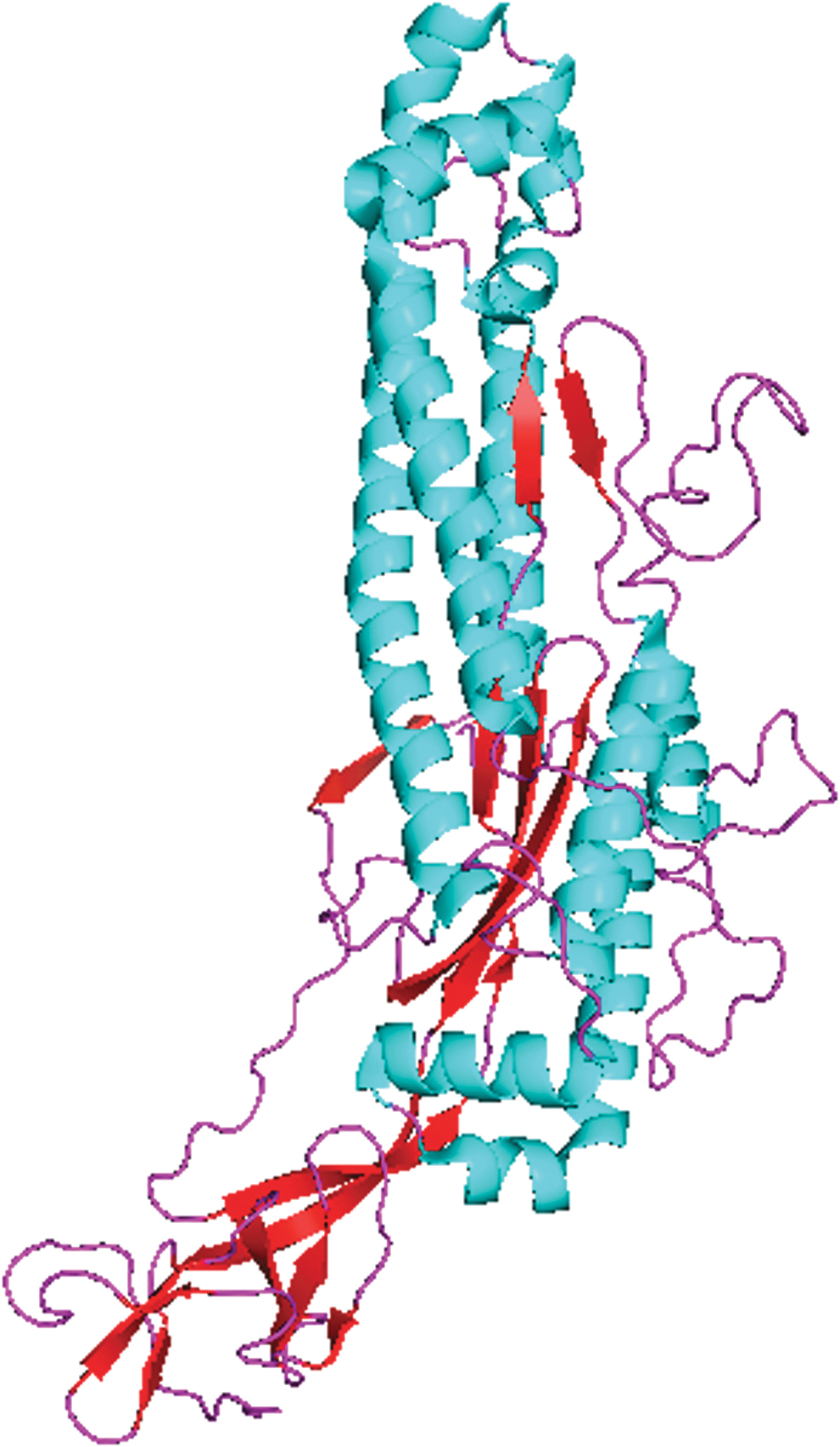
The 3D structure of the S2 domain of the SARS-CoV-2 spike protein. Blue color represents the helix, purple color represents the coil, and red color represents the beta sheets. Color images are available online.
Physicochemical Parameters and Different Properties of the S2 Domain of the Spike Protein of Severe Acute Respiratory Syndrome Coronavirus 2 Computed Through the ExPASy ProtParam Server
GRAVY, grand average of hydropathicity; II, instability index; Mol., molecular.
The transmembrane topology revealed that amino acids 1–528 were present on the surface, amino acids 529–551 were involved in the transmembrane helix, and amino acids 552–588 were inside the S2 domain core. DiANNA predicted the presence of 10 disulfide bonds at different positions (Table 2).
Predicted Disulfide Bonds Within the S2 Domain of the Spike Protein of Severe Acute Respiratory Syndrome Coronavirus 2
Bonds with lowest scores indicated in bold show weak bonds.
Recognition of B cell epitopes
Epitopes were predicted using three methods: the BepiPred linear epitope prediction method 2.0, Emini surface accessibility method, and Kolaskar and Tongaonkar antigenicity method.
The BepiPred linear epitope prediction method revealed 21 peptides, of which only 5 had the ability to act as epitopes. The epitope, LIDLQELGKY, had the highest antigenicity (Fig. 3A) (Supplementary Table S1). By using the Emini surface accessibility method, 11 peptides were predicted. Only four of them were able to generate an immune response. The epitope, DPSKPSKRSF, had the highest antigenicity score (Fig. 3B) (Supplementary Table S1).

B cell epitope prediction of the S2 domain of the SARS-CoV-2 spike protein.
The Kolaskar and Tongaonkar method revealed 17 peptides, of which 7 had an antigenicity score below the threshold value, which means they were nonepitopes. The epitope with the highest antigenicity was TTEILPVS with a score of 1.2071 (Fig. 3C) (Supplementary Table S1).
CTL epitope recognition
A total of 11 epitopes were identified from NetCTL 1.2 (Supplementary Table S2). Antigenicity, toxicity, and immunogenicity of those 11 epitopes were evaluated. All of the epitopes had a length of nine amino acids. Of these, seven epitopes had an antigenicity score below the threshold value, two epitopes were toxic, and six had a negative immunogenicity score.
Of the 11 peptides, only one epitope (RSFIEDLLF) was nontoxic, with a positive immunogenicity score, and its antigenicity score was greater than the threshold value (0.4). Thus, it had the ability to act as a probable epitope and generate an immune response.
HTL epitope prediction
Forty-one epitopes having a percentile rank <10 and length of 15 amino acids were predicted and selected for further assessment (Supplementary Table S3). Almost half of those (20) were nonepitopes. The other half (21) were able to act as epitopes. FAMQMAYRFNGIGVT had the highest antigenicity.
MHC class I epitope prediction
MHC I epitopes were predicted by ProPred-I (Supplementary Table S4). Of the total 39, only 19 epitopes had an antigenicity score below the threshold value (0.4). Rest of the epitopes were nonantigenic. The epitope, NFGAISVVL, had the highest antigenicity score (0.9894), interacting with only two alleles, while the epitope, SAPHGVVFL, was found to interact with the most number of alleles, that is, 9.
MHC class II epitope prediction
ProPred predicted 30 MHC class II epitopes (Supplementary Table S5). Of those, eight were nonantigenic and LPVSMTKTS had the highest antigenic score (1.5550). It was found to interact with almost seven alleles, while the epitope, LLFNKVTLA, was found to interact with the most number of alleles (almost 27).
Eminent feature profiling of MHC class I and class II epitopes
Further properties of selected MHC I and II epitopes were analyzed. Nondigesting enzymes were found through the Protein Digest tool. The epitopes that are digested by fewer enzymes are more stable. ToxinPred was used to distinguish nontoxic epitopes from toxic epitopes. Furthermore, hydrophobicity, hydrophilicity, charge, pI, and mass were also predicted through the ToxinPred tool. These properties can be used to calculate the quality of an epitope.
Results of MHC class I and class II epitopes are given in Supplementary Tables S6 and S7, respectively.
Conservation analysis between different countries
Sequences of the S2 domain of SARS-CoV-2 from eight different variants from reporting countries were downloaded from NCBI. These sequences were forwarded for alignment, and mutations between these sequences were analyzed using Jalview software. Almost 15 mutations were found in the S2 domain of 8 different variants of SARS-CoV-2 (Fig. 4).
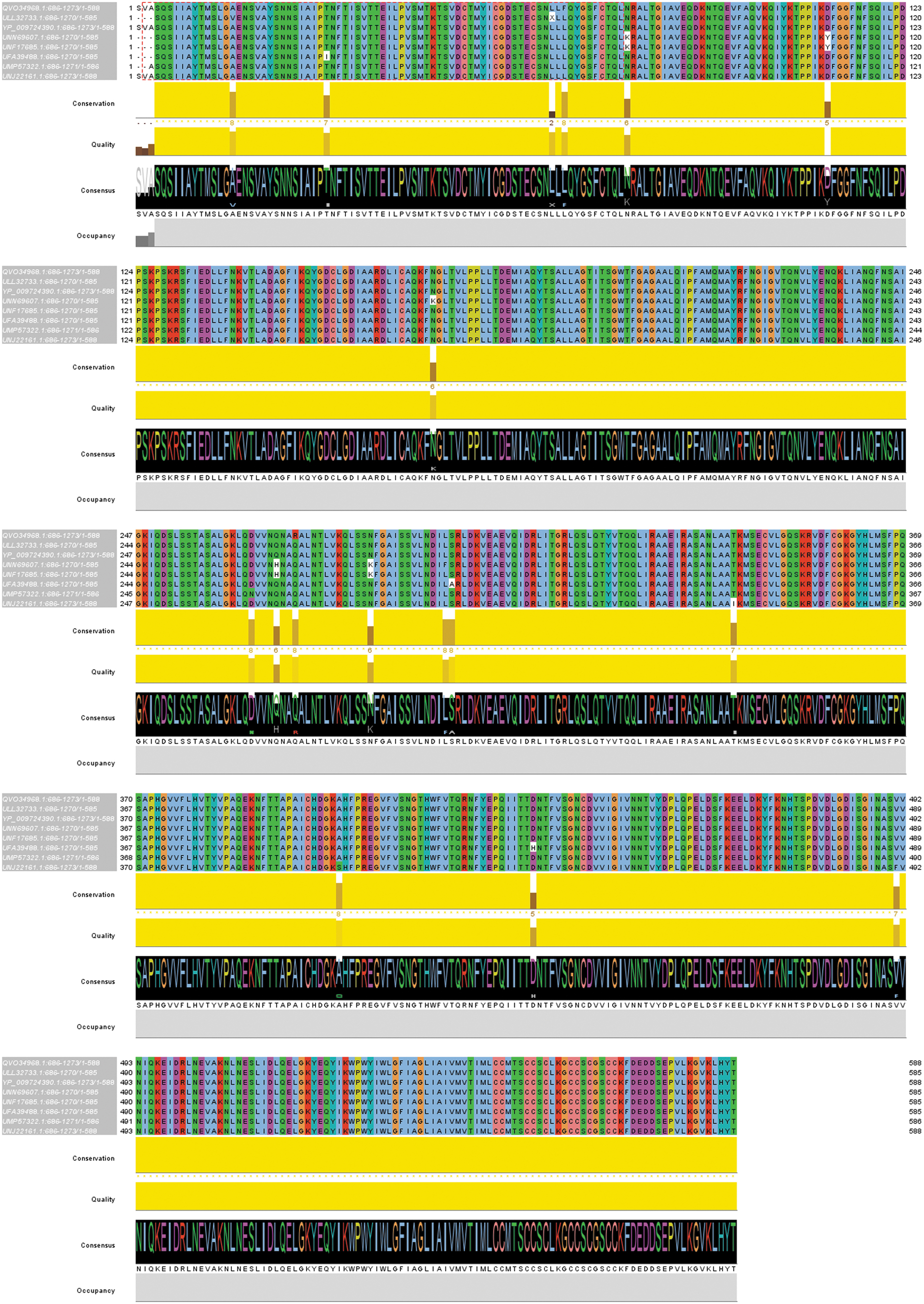
Multiple alignment of different variants of SARS-CoV-2 across the globe. Amino acids shown in white indicate mutations. Fifteen mutations were found in eight different variants of SARSCoV-2. Color images are available online.
A phylogenetic analysis was done to view the conservation between sequences using MegaX. The result showed that the S2 domain among all of the variants of coronavirus was highly conserved (Fig. 5).
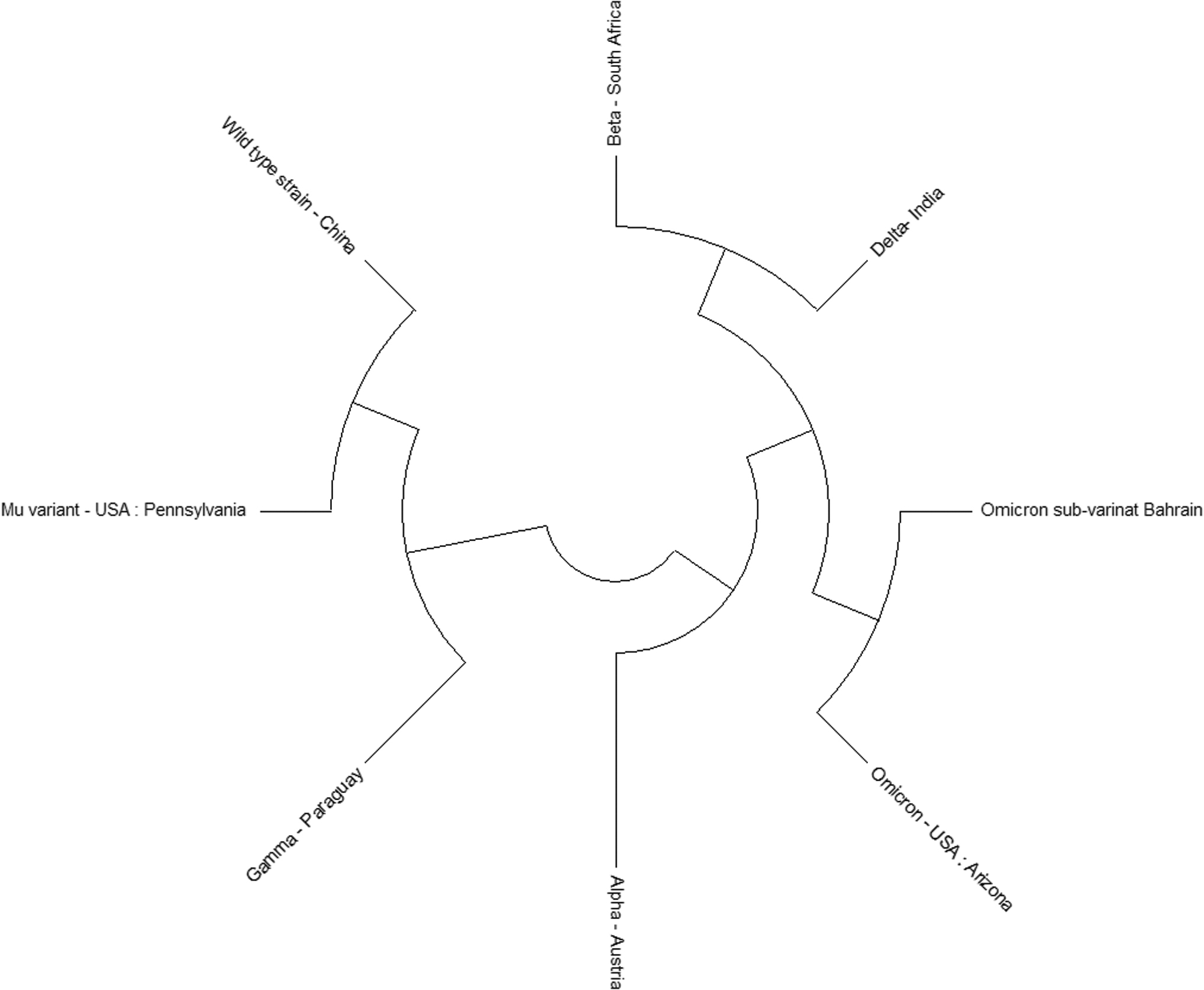
Phylogenetic tree to observe conservation in the S2 domain of the SARS-CoV-2 spike protein among different variants of SARS-CoV-2. Tree shows that the S2 domain is conserved.
Conservation analysis of selected epitopes
Epitope conservancy analysis results revealed that most of the predicted epitopes are from highly conserved regions (Table 3). The epitope, SNLLQYGSFCTQ, with a length of 12 amino acids showed the least conservation score (25%), while most of them appeared to have 100% conservation.
Conservation Analysis of Selected Epitopes of the S2 Domain of the Spike Protein of Severe Acute Respiratory Syndrome Coronavirus 2
Most of the epitopes proved to be conserved.
CTL, cytotoxic T cell lymphocyte; HTL, helper T cell lymphocyte.
Vaccine design
Final epitopes that were predicted in previous steps were joined together with the help of linkers. CTL, HTL and B-cell epitopes were connected with AAY, GPGPG and KK linkers, respectively. The sequence of the final epitope construct is given below. Linkers are shown without highlight. The first light grey highlighted sequence shows CTL epitope, next 21 amino acid sequences joined by the linker “GPGPG” show HTL epitopes, and the next 18 dark grey highlighted amino acid sequences, linked via “KK” linker show B-cell epitopes. The antigenicity of the vaccine was found out to be 0.6136 which shows that this vaccine construct can successfully act as a probable antigen.

Discussion
SARS-CoV-2 rapidly propagated all over the world, causing COVID-19. High transmission and infection rates, rapid mutation in its genome, and unavailability of a vaccine contributed to the quick spread of the pandemic. The common symptoms of COVID-19 were similar to those of common flu caused by the influenza virus. Since the spike protein is present on the outer surface of SARS-CoV-2, it is logical to establish a vaccine construct against this protein.
The spike protein is involved in the receptor recognition process and facilitates entry of the virus. It was also previously thought to initiate a robust immune response in MERS-CoV and SARS-CoV. Epitopes found in the spike protein can be used for construction of the anti-SARS-CoV-2 vaccine (Korber et al, 2020).
Epitopes of the SARS-CoV spike protein were screened and those harboring the same sequence in SARS-CoV-2 were identified (Ahmed et al, 2020) (Grifoni et al, 2020). Despite the sequence homology of 77.38% between spike proteins of SARS-CoV and SARS-CoV-2, antibodies that react against the spike protein of SARS-CoV showed poor response against the SARS-CoV-2 spike protein (Tian et al, 2020). This shows that spike proteins had great genomic diversity.
Although the effectiveness and potential of current vaccines proved beneficial for some time, the potential destruction that it can cause in future should not be overlooked. Among all the vaccines in use, three successfully act against the spike protein. However, since mutations take place more frequently in RNA viruses, it is not a far-fetched hypothesis that these vaccines might not stay totally effective.
The E484K mutation, found in the South African (B.1.351) variant and Brazilian (B.1.128) variant of SARS-CoV-2, is already found to have a negative impact on neutralizing antibodies and thus on vaccine effectiveness (Volz et al, 2021). Most of the in-use vaccines are mRNA based. It was found that both Pfizer and AstraZeneca vaccines showed reduced protectivity against the Beta variant (Chemaitelly et al, 2021; Madhi et al, 2021).
Conservation analysis of the S2 region was done among different variants of SARS-CoV-2. The result showed that the S2 region of SARS-CoV-2 is highly conserved. The same was found previously (Dai et al, 2020). Kuan Cheok Lei and Zhang (2020) found that the N-terminal domain, RBD, and S2 subunit are conserved, but they display different degrees of conservation for different viral strains.
However, Jaiswal and Lee (2022) analyzed the conservation of epitopes of 24 proteins of SARS-CoV-2 and found that 15 epitopes of the S protein, of 106, were conserved in more than 99% of strains. Many mutations are reported in the spike protein gene, which give it the ability to neutralize antibody resistance (McCarthy et al, 2021). These recent mutations are thought to hinder the effect of current vaccines (Wang et al, 2021).
Thus, it is important to study and observe the conservation regions in different variants of SARS-CoV-2 so that in case of any global emergency, it will not be difficult to construct a vaccine. Our work reveals that the S2 region might be a major candidate for development of vaccines in future.
The major advantage of using peptide-based vaccines is induction of target site-specific response. In addition, they also offer excellent alternatives to traditional vaccination approaches because of the ease with which chemical modifications can be introduced (Purcell et al, 2007). The vaccine design of two of the currently used mRNA vaccines is similar. Both use the full-length spike protein stabilized with proline substitutions at two positions (K986P and V987P) and encapsulated in lipid nanoparticles (Pack and Peters, 2022).
BNT162b2 of Pfizer, the first COVID-19 vaccine, was authorized by FDA as the first-ever human mRNA-based vaccine in August 2021 (Parums, 2021). However, a major drawback of these vaccines is their storage requirements. The Moderna vaccine needs a temperature of −20°C for storage up to 6 months and can be stored for 30 days at 2–8°C in the refrigerator, whereas the Pfizer vaccine needs a temperature of −70°C for storage (Ledford, 2020). This cold chain requirement of mRNA vaccines can affect the efficiency of vaccines and can contribute to vaccine waste.
For epitope prediction based on immunoinformatic tools, previously conducted research followed a strategy that is different from this work. Those research works attempted to find epitopes in the whole spike protein, or all the structural proteins, or all the structural plus NSPs. None of the researchers used the S2 domain alone for prediction of suitable epitopes, which showed potential to generate a strong immune response in our study.
The S2 domain is a better site than the whole spike protein for development of a multiepitope peptide vaccine (MPV) because it will allow focus only on the immunogenic parts of the protein and ignore features that are less important in triggering an immune response.
A study revealed that 6 of 20 T cell epitopes of the spike protein were present in the S2 region (Zhao et al, 2021). Another study also reported that high IgG-mediated antiviral protection was also induced by the S2 region (Walls et al, 2020). Devi et al (2021) found 11 epitopes for the S protein and 1, 2, and 6 epitopes for E, M, and N proteins, respectively.
Numerous studies have now reported neurologic, cardiac, and other side effects after the use of currently available SARS-CoV-2 vaccines, which use the whole spike protein to induce an immunogenic response (Di Resta et al, 2021; Finsterer, 2022; Ishay et al, 2021; Menni et al, 2021; Shiravi et al, 2022). Deducing from the fact that the functional capacity of a protein is based on its structural integrity, we assume that using its domain might minimize the risks of allergic or autoimmune reactions to the whole spike protein.
In the hunt for a multiepitope-based vaccine against SARS-CoV-2, Safavi et al (2020) conducted a study. They selected six NSPs and the immunodominant part of the spike protein of SARS-CoV-2 for predicting the vaccine construct. In comparison with the mentioned study, our study also included a conservation analysis of different variants of SARS-CoV-2. It could provide a better understanding in terms of monitoring mutations that have occurred so far in the SARS-CoV-2 spike protein region. Moreover, the conservation analysis can also be useful in future studies for observing and comparing the conserved and nonconserved regions in different variants.
The research by Safavi et al for constructing vaccines based on epitopes was conducted in 2020. More data are now available for a better understanding of the viral genome, and the predicted epitopes need updating accordingly. Moreover, after introduction of the vaccines in 2020, the virus followed a natural course of survival by mutating and developing resistance mechanisms. As the data have grown immensely over the course of these two and a half years, an updated epitope design will increase confidence in the final product.
HTL and CTL epitopes were predicted because these are important intermediates of cellular immunity. Only one CTL epitope, RSFIEDLLF, based on its toxicity, antigenicity, and immunogenicity properties exhibited the potential for vaccine development. However, for HTLs, the epitopes passing all filters were 21 in number. Of those 21 epitopes, FAMQMAYRFNGIGVT with a length of 15 aa had the highest antigenic score (1.3688), while REGVFVSNGTHWFVT with a length of 15 aa had the lowest antigenic score (0.4461).
The B cell epitope analysis resulted in 18 epitopes with predicted potential to generate an immune response. Almost 90% of the selected epitopes showed 100% conservation. This indicates that these peptides meet the prerequisites for peptide vaccines. Two of the predicted B cell epitopes, VPAKEQNFT and TTEILPVS, overlapped with T cell epitopes, VFLHVTYPAQEKNF and NFTISVTTEILPVSM, respectively. This shows that both these epitopes were involved in B cell and T cell immune activation at the same time.
Successive research work is needed to validate the effectiveness of our work in in vitro and in vivo models. Mahdevar et al (2022) also adopted an immunoinformatic-based approach to predict a vaccine against breast cancer and selected the BORIS cancer-testis antigen for this purpose. They also selected epitopes for vaccine construction on the basis of antigenicity score. The antigenicity score of their final vaccine construct was 0.5286, whereas the antigenicity score revealed by the VaxiJen 2.0 server was 0.6136 for our vaccine's final construct. Their final vaccine construct's efficacy was determined by checking its effect in vivo.
Antitumor effects of MPV were evaluated in the nonimmunogenic 4T1 mammary carcinoma in BALB/c mice (Mahdevar et al, 2021). Results showed that the vaccine not only successfully decreased the tumor's size but it also inhibited tumor growth and increased the survival time of tumor-bearing mice.
Results also showed that peptide vaccine immunization significantly increased production of antibodies, interleukin-4 (10 fold) and interferon-γ (16 fold) (Safavi et al, 2019). Our vaccine construct's antigenicity score is better than Mahdevar et al's, which helps us conclude that it can also enter in vivo studies successfully.
Conclusions
The S2 domain of SARS-CoV-2 has shown strong potential for CTL, HTL, MHC I, MHC II, and B cell epitope construction. Furthermore, the conservation analysis performed on different variants of SARS-CoV-2 revealed that the virus mutated gradually. As some of the in-use vaccines are not so effective against some mutants of SARS-CoV-2, there is still space for further improvement of antiviral therapy and development of an effective drug and vaccine to fight the ongoing pandemic.
The S2 domain might be a strong candidate for mRNA-based vaccine constructs given the reported side effects of using the whole spike protein in the vaccine.
Footnotes
Acknowledgments
The authors would like to thank the chief librarian and staff of the Superior University Library, main campus, for providing the internet and proper seating facilities to conduct this research.
Authors' Contributions
F.N. conducted the analysis, wrote the first draft, and formatted it. R.N. conceived, supervised, and wrote the final draft. A.A., A.A., S.A., and M.S. performed and analyzed different steps of the pipeline. A.S., A.N., and U.M. helped design the first draft and result analysis. M.I. supervised the whole study.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.