
Abstract
Emerging severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) variants have raised concerns about the efficacy of vaccines. The present study aimed to compare the potential of Delta and Omicron variant-specific mRNA vaccines in inducing immune responses. B cell and T cell epitopes and population coverage of spike (S) glycoprotein of the variants were predicted using the Immune Epitope Database. The molecular docking was carried out between the protein and different toll-like receptors, as well as the receptor-binding domain (RBD) protein and angiotensin-converting-enzyme 2 (ACE2) cellular receptor using ClusPro. The molecular simulation was done for each docked RBD-ACE2 using YASARA. The mRNA secondary structure was predicted through the RNAfold. The simulation of immune responses to the mRNA vaccine construct was performed using C-ImmSim. Apart from a few positions, no significant difference was observed in the prediction of S protein B cell and T cell epitopes of these two variants. The lower amounts of Median consensus percentile in the Delta variant in similar positions signify its stronger affinity to major histocompatibility complex (MHC) II binding alleles. Docking of Delta S protein with TLR3, TLR4, and TLR7 and also its RBD with ACE2 showed striking interactions with the lower binding energy than Omicron. In the immune simulation, elevated levels of cytotoxic T lymphocytes, helper T lymphocytes, and memory cells in both the active and resting states and the main regulators of the immune system suggested the capacity of mRNA constructs to elicit robust immune responses against SARS-CoV-2 variants. Considering slight differences in the binding affinity to MHC II binding alleles, activation of TLRs, mRNA secondary structure stability, and concentration of immunoglobulins and cytokines, the Delta variant is suggested for the mRNA vaccine construction. Further studies are being done to prove the efficiency of the design construct.
Introduction
The recent COVID-19 pandemic caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) affected the health of millions of people worldwide. All of the developed vaccines have focused on induction specific antibody responses against virus spike (S) glycoprotein. The surface-exposed location of S protein makes it play a major role in viral infection, elicit humoral immune and protective cellular immune responses, and is the primary target of neutralizing antibodies (Shang et al, 2020; Walls et al, 2020). The S glycoprotein is synthesized as a 1,273-amino acid polyprotein precursor proteolytically cleaved by ubiquitous cell proteases at the S1/S2 cleavage site. The surface subunit S1 binds to the angiotensin-converting-enzyme 2 (ACE2) receptor and triggers viral entry following virus-cell membrane fusion, which mediates by the transmembrane subunit S2 (Zhang et al, 2020).
Following viral entry, the genomic RNA is released into the host cell cytoplasm and translates into viral replicase polyproteins (pp1a and 1ab). The polyproteins are further cleaved by viral proteases into 16 nonstructural proteins and form a replication–transcription complex. The complex mediates genome replication and transcription process of nucleocapsid (N) structural protein, which is synthesized in the cytoplasm and also other viral proteins, including S protein, membrane (M) protein, and envelope (E) protein that is translated in the rough endoplasmic reticulum then transported to the Golgi complex. In the trans-Golgi network, the S glycoprotein is subjected to post-translational processing and cleavage. Interaction between the S1 receptor-binding domain (RBD) and ACE2 is essential for tissue tropism and pathogenesis of the infection (Kim et al, 2020; V'kovski et al, 2021). During virus–cell interaction, SARS-CoV-2 genomic RNA and structural components are recognized by pathogen-associated molecular patterns, which continue throughout the viral lifecycle in a host cell.
Upon infection, a series of pattern recognition receptors are activated, and the innate immune response is triggered through the toll-like receptor (TLR). The toll-interleukin-1 receptor (TIR) domain of surface and endosomal TLRs recruits adaptor proteins resulting in downstream signaling of generating the pro-inflammatory cytokines and interferon (IFN) responses (Madden and Diamond, 2022). The humoral response involves specific neutralizing antibodies mainly against the S protein, precisely the receptor-binding motif (RBM) in the RBD region. The S gene presents a high mutational range critical for virus infectivity and transmissibility (Harvey et al, 2021). Several factors mainly the incomplete or insufficient control measures increase the emergence of variants, which have increased pathogenicity, ACE2 receptor binding affinity, reinfection, and immune escape risks (De Masi et al, 2022). From the first identification of SARS-CoV-2 in Wuhan of China in 2019, the emerging variants are classified as variant under investigation, variant of interest, or variant of concern (VOC).
Changes in ACE2 receptor affinity, increase in virus load in infected cells, greater transmissibility, immune escape, and risk of reinfection are characteristics of VOCs that are obtained through selective or survival benefits (Harvey et al, 2021; Tao et al, 2021). Among them, the B.1.617.2 (Delta) variant identified in Maharashtra of India in late 2020 is of great concern to the world due to the high prevalence and transmissibility with widespread fatalities. The Delta variant possesses the synonymous amino acid substitutions, which confer increased infectivity and a relative loss of susceptibility to neutralizing antibodies (Mlcochova et al, 2021; Planas et al, 2022). Compared to the dominant variant Delta, Omicron (B.1.1.529; first reported from southern Africa in November 2021) carries more mutations, especially in RBM that may significantly impact the binding affinity to ACE2 receptor and transmissibility (Karim and Karim, 2021; Wu et al, 2022).
From early 2020 till now, different kinds of vaccines, including inactivated vaccines, mRNA vaccines, and subunit protein vaccines constructed within carrier vectors, are globally used against SARS-CoV-2. Although the idea of designing and developing an mRNA vaccine was launched in Dr. Robert Malone's laboratory in late 1987, vaccines based on the technology were approved for use in the recent SARS-CoV-2 pandemic in early 2020 (Dolgin, 2021). Given the unavoidable advantages of the mRNA vaccine over other vaccine platforms, we compared the capabilities of Delta and Omicron mRNA constructs in inducing immune responses against SARS-CoV-2 with the aim of providing insights into the impact of new variants on the effectiveness of current vaccines.
Materials and Methods
Retrieval of S glycoprotein sequence
The FASTA formatted nucleotide and amino acid sequences of S glycoprotein from the Delta and Omicron variants (OM142697 and OM066778) were retrieved from the Virus Pathogen Database and Analysis Resource (ViPR,
Linear B cell epitope prediction
B lymphocytes are responsible for humoral immunity and long-term immunological protection by secreting antigen-specific antibodies. The S protein sequences were screened for linear continuous B cell epitopes using Immune Epitope Database (IEDB) (Vita et al, 2019). The threshold ≥0.5 was the main parameter for the sequential residues predicted to be B cell epitopes. The sequences were predicted for antigenicity using Kolaskar and Tongaonkar's algorithm.
T cell epitopes and population coverage prediction
A protective vaccine should induce specific T cell responses based on major histocompatibility complex (MHC)-presented peptides. Potential cytotoxic T lymphocyte (CTL) epitopes binding to MHC class I were predicted using TepiTool available in IEDB. A panel of 27 most frequent HLA-A and -B alleles was selected to identify the CTL epitopes using the NetMHCpan method, which deploys an artificial neural network (ANN)-based methodology to execute epitope prediction. At a cutoff of IC50 ≤ 50, the strong 9-mer MHCI binders were predicted. The seven allele method was used for prediction of MHC class II-binding helper T lymphocyte (HTL) epitopes with a median consensus percentile rank ≤20. To determine the vaccine's coverage, population coverage of the predicted epitopes and their respective MHC HLA-binding alleles were analyzed using the IEDB population coverage tool.
Physicochemical properties and allergenicity evaluation
Physicochemical properties, including molecular weight, theoretical isoelectric point (pI), instability index, aliphatic index, and grand average of hydrophobicity (GRAVY), were detected using ProtParam at the ExPASy online web server. The aliphatic index is the positive factor for the increase in thermostability of a protein. Hydrophobicity property displays the possibility of being a globular (hydrophilic) protein that was indicated by the GRAVY range below 0. Allergenicity was evaluated through AllerTOP v. 2.0 using the k-nearest neighbor (kNN, k = 1). Based on the algorithm the protein sequence is classified to allergens and/or nonallergens.
Vaccine designing procedure
In mRNA vaccine construction five key elements that include 5′ and 3′ untranslated regions (UTRs), methylated 5′ cap (m7Gp3N), poly (A) tail, and Kozak sequence are critical for increasing the life span and expression of the mRNA molecule (Verbeke et al, 2021). We used AntaRNA with predefined nucleotide composition and minimal secondary structure to design the 5′UTRs. 3′UTR sequence was detected using 3USS. The secondary structure of the vaccine constructs was predicted using SOPMA online server. Then the tertiary (three-dimensional [3D]) structure models were built using SWISS-MODEL and validated. The potential antigenicity was screened through VaxiJen v2.0 server.
Molecular docking and molecular dynamics simulation
To ensure the efficacy of the vaccine's construct in inducing the immune responses, binding affinity and orientation between the construct and cellular ligand were predicted using ClusPro v2.0. Among TLRs, activation of TLR7 and potentially TLR3 and TLR4 is critical for priming immune responses. The refined 3D model of the vaccine constructs was docked against human TLR7 (PDB ID: 7CYN), human TLR3 (PDB ID: 1ZIW), and human TLR4 (PDB ID: 4G8A). Based on the algorithm, the lower calculated energy score correlates with better binding affinity (Kozakov et al, 2017). Binding affinity and stability of the docked complexes based on pair-wise intermolecular contacts within 5.5Å distance threshold were determined using PRODIGY. Different types of interaction such as electrostatic, van der Waals, and polar were calculated by Gibbs free energy (ΔG) and dissociation constant (Kd) at 25°C.
To evaluate the interaction of the virus variants S protein with the human ACE2, molecular docking was performed between the RBD protein and this cellular receptor (PDB ID 1R42). The docked complex molecular dynamics (MD) simulations were evaluated through root-mean-square deviation (RMSD) from the trajectory of 100 nanoseconds (ns) simulation using YASARA dynamic software.
Codon adaptation and mRNA secondary structure analysis
Translation efficiency of the mRNA constructs was assayed using the JCat. Codon sequence was optimized for Escherichia coli K12 expression system, and GC content and Codon Adaptation Index (CAI) were calculated. The mRNA secondary structure was predicted through the RNAfold server to check the translation efficiency and thermodynamic stability.
Immune response simulation
The in silico simulation of humoral and cell-mediated immune responses to the mRNA vaccine construct was carried out using C-ImmSim server (
Results
Prediction of B cell epitopes
The most significant linear B cell epitopes for S protein of Delta and Omicron variants were identified using BepiPred 2.0 (Table 1). With slight differences, the positions and epitope sequences of the protein were the same in both variants. These differences corresponded to the deletions at positions 69–70, 143–145, and 211 and the insertion at 214. Kolaskar and Tongaonkar's algorithm estimated the average antigenicity 1.041 (Max 1.261 and Min 0.866) for both variants. The data indicated mutations in S protein of Omicron did not alter its antigenicity compared to Delta variant.
B Cell Epitope Predictions of Severe Acute Respiratory Syndrome Coronavirus-2 Spike Protein of Delta and Omicron Variants
Epitopes that are not predicted in a variant at the same position are marked with bold NO, which indicates the difference.
NO, not predicted.
Identification of CTL and HTL epitopes and population coverage
CTL epitopes of 9-mer in length and IC50 < 50 for S protein were predicted through binding affinity with the most frequent human alleles (Table 2). Apart from the five positions, no significant difference was observed in the prediction of S protein epitopes of these two variants. The epitope at position 491 of the Omicron variant showed an affinity to bind to seven HLA alleles, whereas this epitope was not predicted for the Delta variant. This difference was observed in the binding affinity at position 976 of the Delta variant or 971 of the Omicron. The HTL epitopes were predicted by the seven allele method (Table 3). In different positions, four epitopes in the Delta and four epitopes in the Omicron were not detected. In similar positions, the amount of Median consensus percentile in the Delta variant was lower, which means a stronger affinity to MHC II binding alleles. The population coverage analysis was done for S protein epitopes having strong binding affinity and their respective CTL and HTL alleles. The combined coverage was estimated at 95.6% with average hit of 10.58 of global population for both variants (Fig. 1).
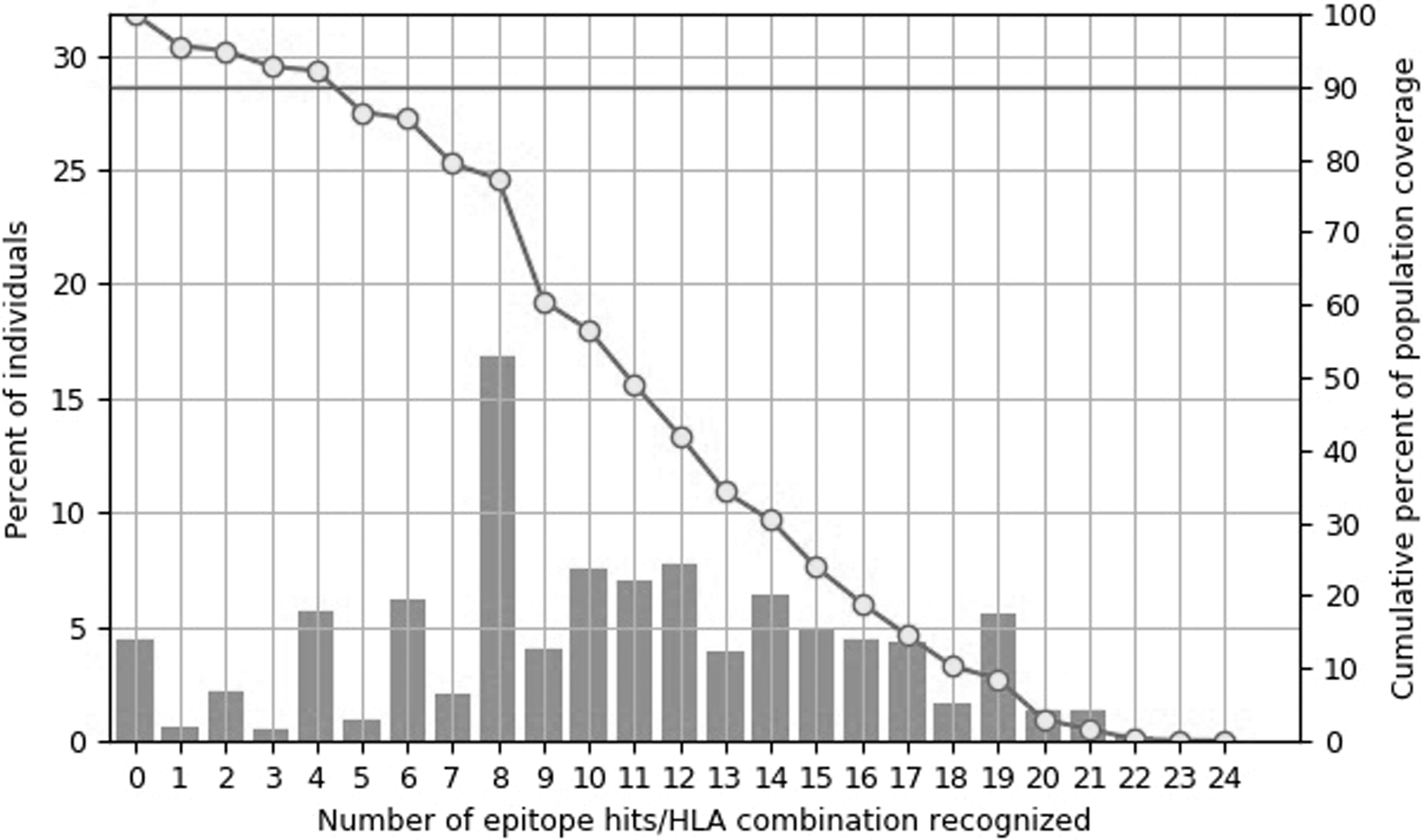
Global population coverage analysis of the strong binding B cell and T cell consensus epitope sequences computing based on genotypic frequencies of MHC I and MHC-II HLA binding alleles. MHC-I, major histocompatibility complex I.
The Major Histocompatibility Complex I Binding Predictions of Severe Acute Respiratory Syndrome Coronavirus-2 Spike Protein of Delta and Omicron Variants to the Most Frequent HLA-A and -B Alleles
Epitopes that are not predicted in a variant at the same position are marked with bold NO, which indicates the difference.
The cutoff for the peptide selection was <50.
The Major Histocompatibility Complex II Binding Predictions of Severe Acute Respiratory Syndrome Coronavirus-2 Spike Protein of Delta and Omicron Variants to the Most Frequent DR Alleles Based on 7-Allele Method
Epitopes that are not predicted in a variant at the same position are marked with bold NO, which indicates the difference.
The median consensus percentile selection was <20.
Evaluation of physicochemical properties
Due to changes in the amino acid composition of S protein in Delta and Omicron variants, its physicochemical properties were analyzed. The S proteins were found to be basic in nature by identifying the PI of 6.94 for Delta and 7.54 for Omicron. The estimated half-life was calculated 30 h in mammalian reticulocytes, >20 h in yeast and >10 h in E. coli. Other properties, including GRAVY (−0.091 and −0.090), aliphatic index (84.14 and 84.35), and instability index (33.19 and 34.61), were very similar for these variants. The estimated negative values of GRAVY indicated that the protein which is hydrophilic can interact with water molecules. The aliphatic index is the positive factor for the increase in thermostability of a protein. In this study, the high index indicated that the protein is thermostable over a wide temperature range. In addition, the protein was defined as nonallergen.
Validation of vaccine construct
The secondary structure of the constructs was analyzed and revealed a flexible and stable structure for both variants. Delta has a higher fraction of alpha-helix structure than Omicron (28.35% vs. 27.94%) and slightly increased in random coil composition (46.69% vs. 46.01%), but less extended strand structure (21.35% vs. 22.44%).
The 3D structure of Delta vaccine construct was modeled based on template 7wg9.1 with a high sequence identity 99.76%, GMQE score 0.71, and QMEAN Z score −3.95. The best model for Omicron based on template 7we7.1 had 98.50% sequence identity, GMQE score 0.71, and QMEAN Z score −2.36. According to SWISS-MODEL documentation, the number between 0 and 1 for GMQE score reflects the expected accuracy of the model and coverage of the target, and the QMEAN Z score greater than −0.4 provides an estimate of the degree of nativeness of the structural features observed in the model on a global scale. Validity of these structures was analyzed by Ramachandran plot and shows that more than 93% of the amino acid residues were within the most favored regions; about 5% in allowed regions; and 2% in outliers regions. The negative Z-score of ProSA, −4.15 for Delta and −3.47 for Omicron, showed high matching between the 3D modeled structures and the related templates. These data denote a high accuracy rate for the modeled constructs.
These sequences were also screened out using VaxiJen for confirmation of antigenicity at threshold of 0.2. With prediction of 0.4735 and 0.4802 scores for Delta and Omicron, both were identified as antigenic.
Molecular docking and MD
An efficient immune response is produced if the vaccine construct properly interacts with the human immune receptor. Therefore, the interaction was analyzed by binding affinity between the vaccine construct and TLRs. The molecular docking was performed through the ClusPro server, which uses energy based filtering and clustering property algorithms for ranking the docked structures. In this study 30 clusters were generated for each vaccine peptide and TLR docking complex. Based on thermodynamics, the best cluster had lowest energy score and the highest binding affinity between the construct and TLR. Energy scores calculated for TLR3-, TLR4-, and TLR7-Delta complexes were −1,296.1, −11.89.6, and −1,190.9 kcal/mol and for Omicron were −1,235.0, −1,205.7, and 1,058.2 kcal/mol, respectively. The larger negative potential energies docked complexes underlined the greater conformational flexibilities of these proteins. As a result, the relative higher binding interaction of Delta with all of the TLRs allows for more developing of innate and adaptive immunity than Omicron.
The interface interactions and energies were also calculated by PRODIGY and compared. The ΔG for binding interaction of Delta and TLR complexes was predicted as −12.6 kcal/mol for TLR3, −11.7 kcal/mol for TLR4, and −9.8 kcal/mol for TLR7. The Kd value of complex dissociation constant was calculated as 3.4E-9 M, 2.7E-9 M, and 2.9E-08 M, respectively. The ΔG for Omicron and TLR complexes was predicted as −11.8 kcal/mol for TLR3, −11.2 kcal/mol for TLR4, and −10.6 kcal/mol for TLR7. The Kd value was calculated as 5.2E-09 M, 4.8E-09 M, and 4.2E-08 M, respectively, which is higher than those observed in the complexes with Delta. Although the binding affinity is greater in the smaller Kd, such little differences in the value probably have no effect on the induction of immune responses.
RBD protein of both variants was bound to ACE2 with the lowest binding energy of −997.2 kcal/mol for Delta and −920.2 for Omicron. Two main regions consisting of shallow grooves on the ACE2 are involved in the interaction; one region is formed by E37, K353, G354, and D355 and the other by D30 and K31. Different amino acid residues in the RBD of Delta and Omicron allow them to bind differently with ACE2. For Delta RBD protein, the residues K417, L455, E484, P491, and Q493 interact with D30 and K31 region, and the residues R403, Y449, Y453, N 487, Y489, G496, F497, Q498, T500, G502, Y505, and Q506 interact with the other region on ACE2 (Fig. 2). At least four RBD Omicron mutations change the binding affinity to ACE2 compared to Delta. Substitutions K to N at position 417, E to A at 484, and G to S at 496 resulted in the decrease in binding affinity to ACE2 at D30, D38, L45, L79, and K353 residues, while the substitution Q to K at position 493 increases the binding affinity to ACE2 at K31, H34, and E35.

Binding affinity of human ACE2 to the SARS-CoV-2 RBD spike protein on the three-dimensional structure. RBD protein is represented as backbone ribbons. Arrows represent ACE2 amino acids that do not interact with the Omicron RBD. ACE2, angiotensin-converting-enzyme 2; RBD, receptor-binding domain; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2.
To analyze the effect of mutation on the ACE2 receptor binding stability of the RBD protein, MD simulation was performed for a 100 ns timescale and the RMSDs were plotted (Fig. 3). The Delta RBD-ACE2 complex showed an almost constant deviation of about 2.0Å reflecting the stable interaction between the RBD protein and receptor molecule. Despite no remarkable change being observed between these variants at the 20 ns, the value of the Omicron variant became increased up to ∼4.0Å during the simulation period. The RMSD fluctuations had ∼2Å differences for these complexes, and the Omicron docked RBD-ACE2 showed higher fluctuations indicating less stability than the Delta.
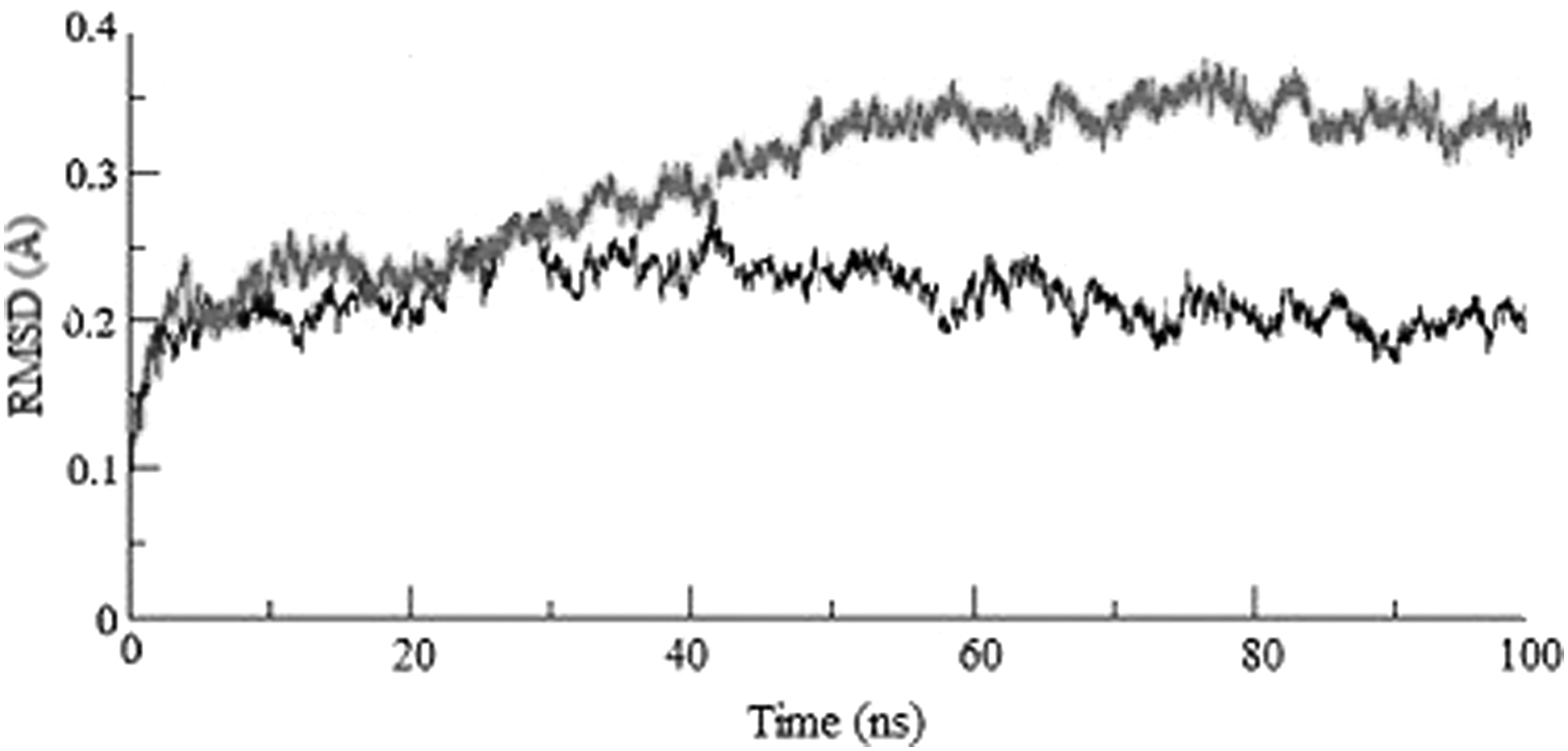
The stability of the RBD-ACE2 docked complexes was evaluated using the Root Mean Square Deviation (RMSD) plot for the Delta (down) and the Omicron (up) over 100 nanoseconds (ns) simulation as a function of time. RMSD, root-mean-square deviation.
Codon optimization and analysis of mRNA secondary structure
GC contents of optimized nucleotide sequence were 50.69% for Delta and 50.48% for Omicron. The calculated CAI values were 0.934 and 0.937, respectively. The GC content ranging between 30% and 70% and a CAI value greater than 0.8 up to 1 are considered optimal for better expression of gene construct.
In thermodynamic ensemble prediction using RNAfold server, the minimum free energy (MFE) of mRNA secondary structure was −1,453.99 kcal/mol for Delta and −1,435.91 kcal/mol for Omicron. As expected amino acid substitutions affected the RBD mRNA secondary structure (Fig. 4). The mutations in Omicron RBD formed more pseudoknot structures upon base pairing in the mRNA compared to Delta RBD, which can affect the protein binding, tertiary folding, and stability. Due to the mutations, the calculated RBD MFE was lower in Delta than Omicron (−124.21 kcal/mol vs. −119.81 kcal/mol). The difference in the energy level indicates that the mRNA secondary structure of Delta is more stable compared with Omicron, which is confirmed by the insertion of pseudoknot structures.
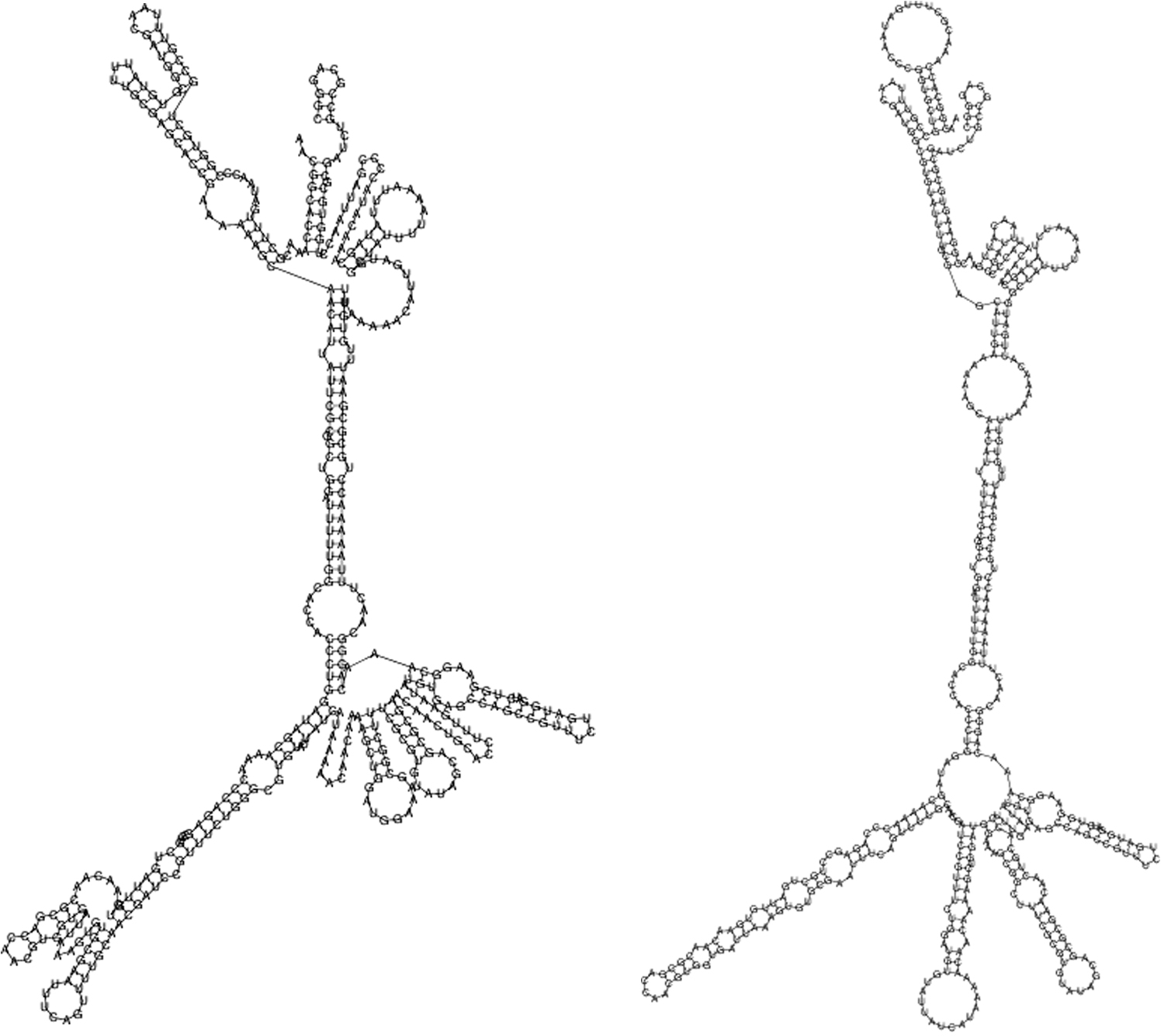
RBD mRNA secondary structure of Delta (left) and Omicron (right) variants. Nucleotide mutations especially in the binding motif resulting in insertion of pseudoknot structures (hairpin loop, bulge loop, and interior loop) and change in the MFE level. MFE, minimum free energy.
Immune simulation
The immune response elicitation of the vaccine constructs was examined using the C-ImmSim server for 100 simulation steps (Fig. 5). The minimum time interval between taking two doses was considered 3 weeks. Interpretation of the simulation results revealed the development of immune response after immunization with both variants; however, the secondary response is much higher than the primary response following the injection of two doses of these vaccines. The IgG + IgM and IgG1 antibody concentrations were higher than those of other immunoglobulins (Fig. 5A). B cell subpopulations, including B cell memory and plasma B lymphocytes, were highly stimulated upon immunization (Fig. 5B). In particular, the association of active B cells and memory B cells with the production of higher affinity antibodies such as IgG was following the first injection (days 10–20). The isotype levels were entirely lower for Omicron.
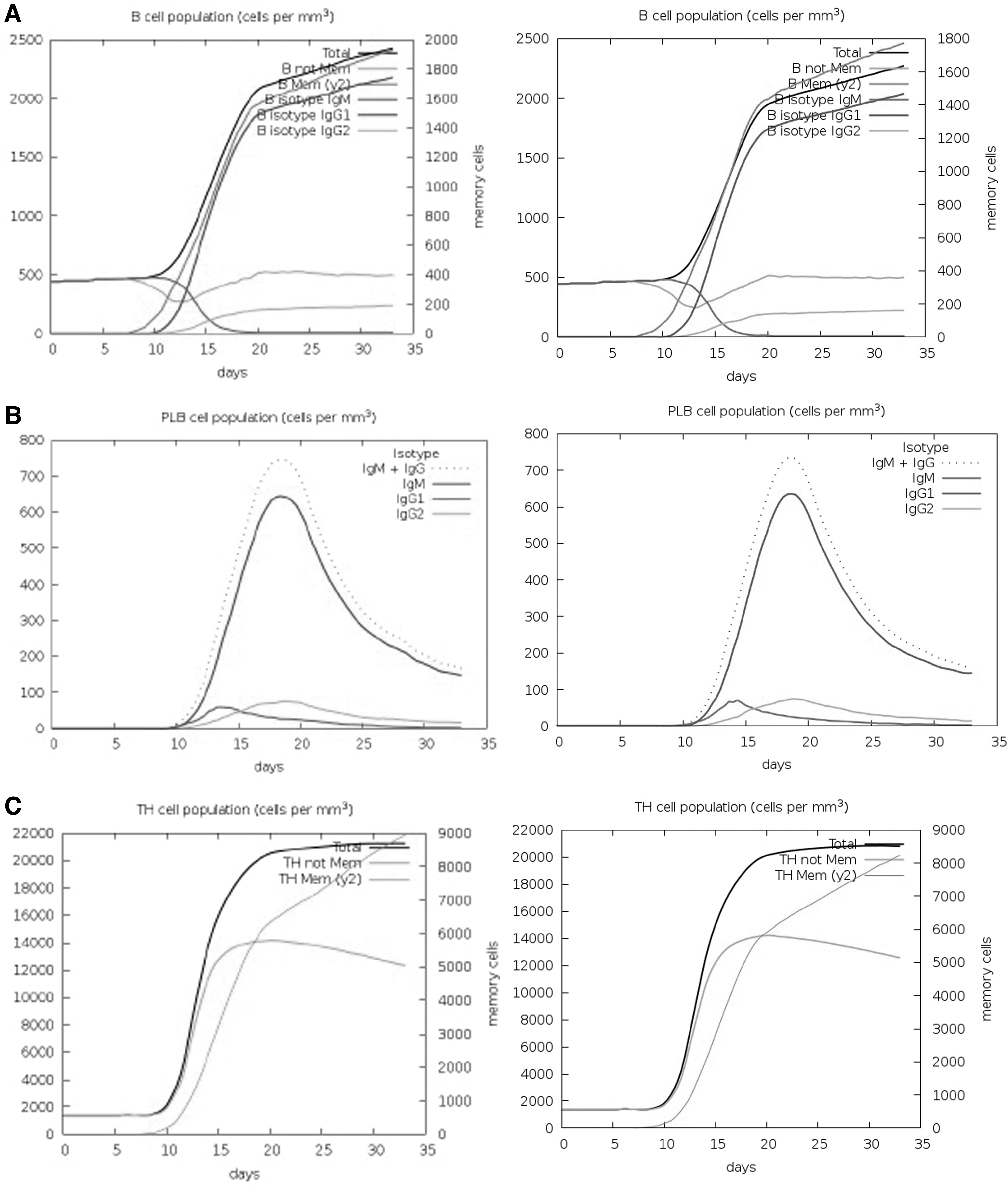
C-ImmSim presentation of computational immune simulation of the Delta and Omicron variants.
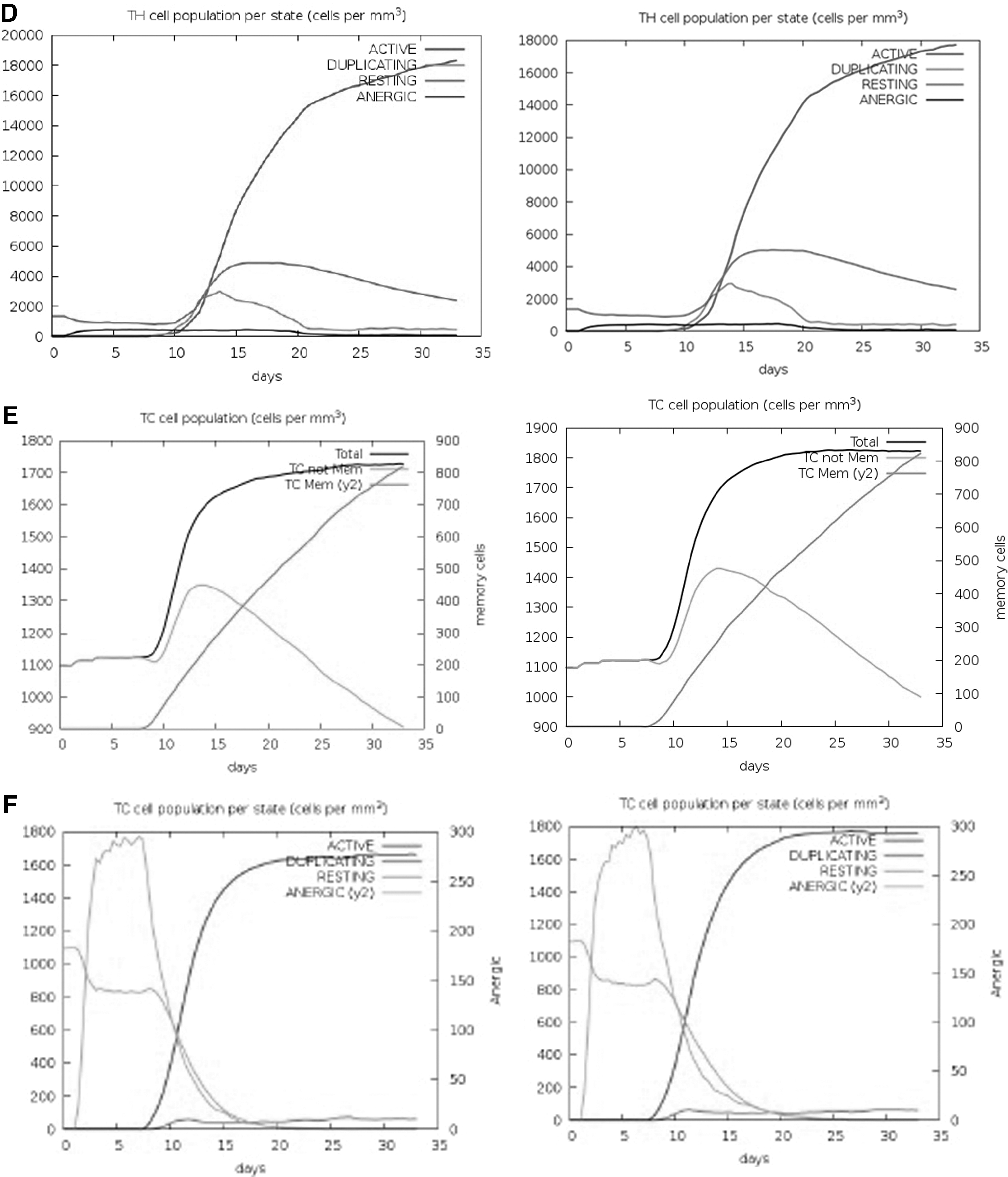
The delay in the increase in B cell levels was observed, while T cell subsets especially CTLs increased during the consecutive days 12–35 for both variants. T helper cells showed very promising results and increased on day 10 to a level of 15,000 cells/mm3 and elevated to 18,000 up to 35 days consequently evoking a high number of memory cells (Figs. 5C, D). Elevated levels of active cytotoxic cell populations and an increased level of memory cells were also observed throughout the stimulation period (Fig. 5E, F).
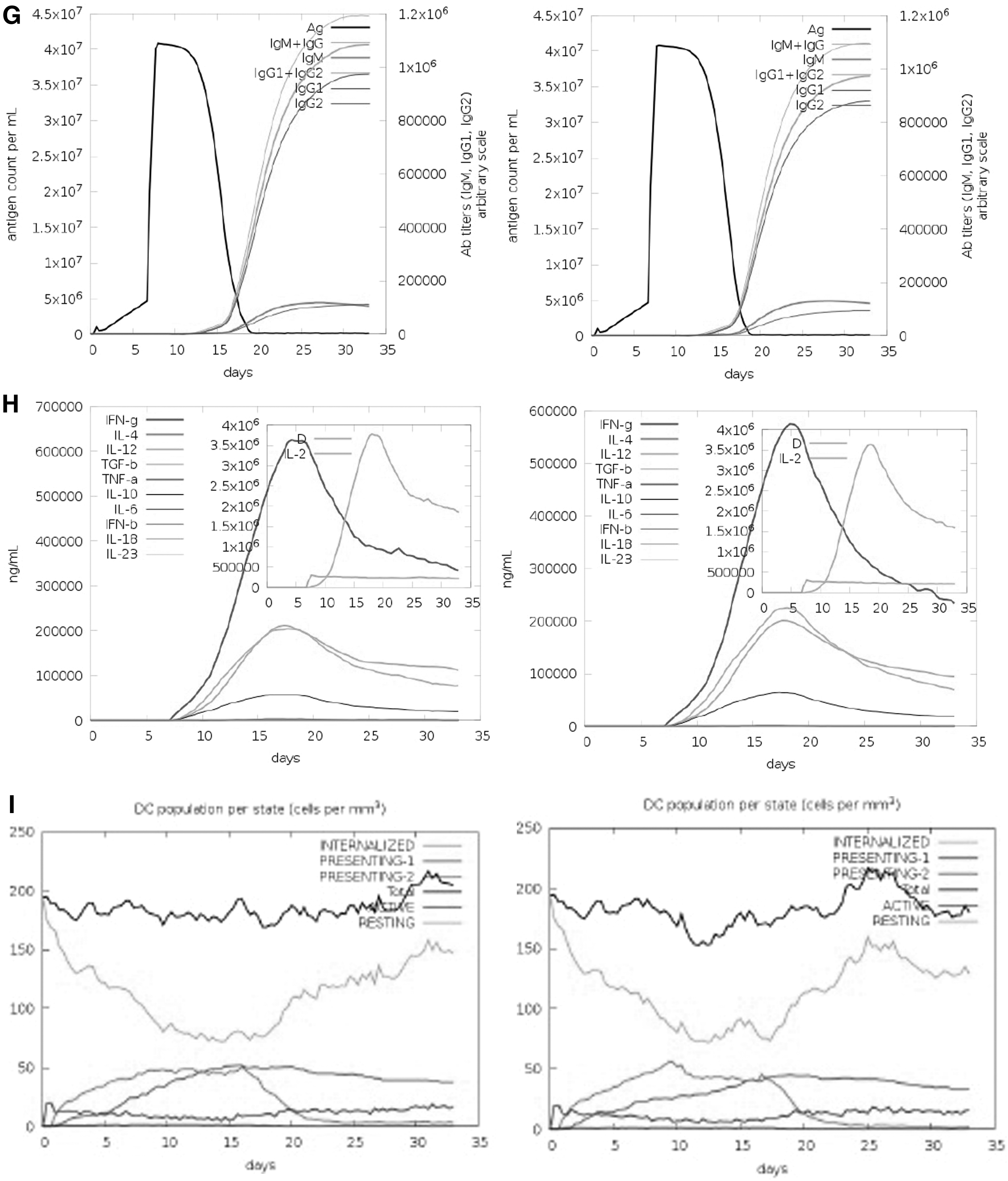
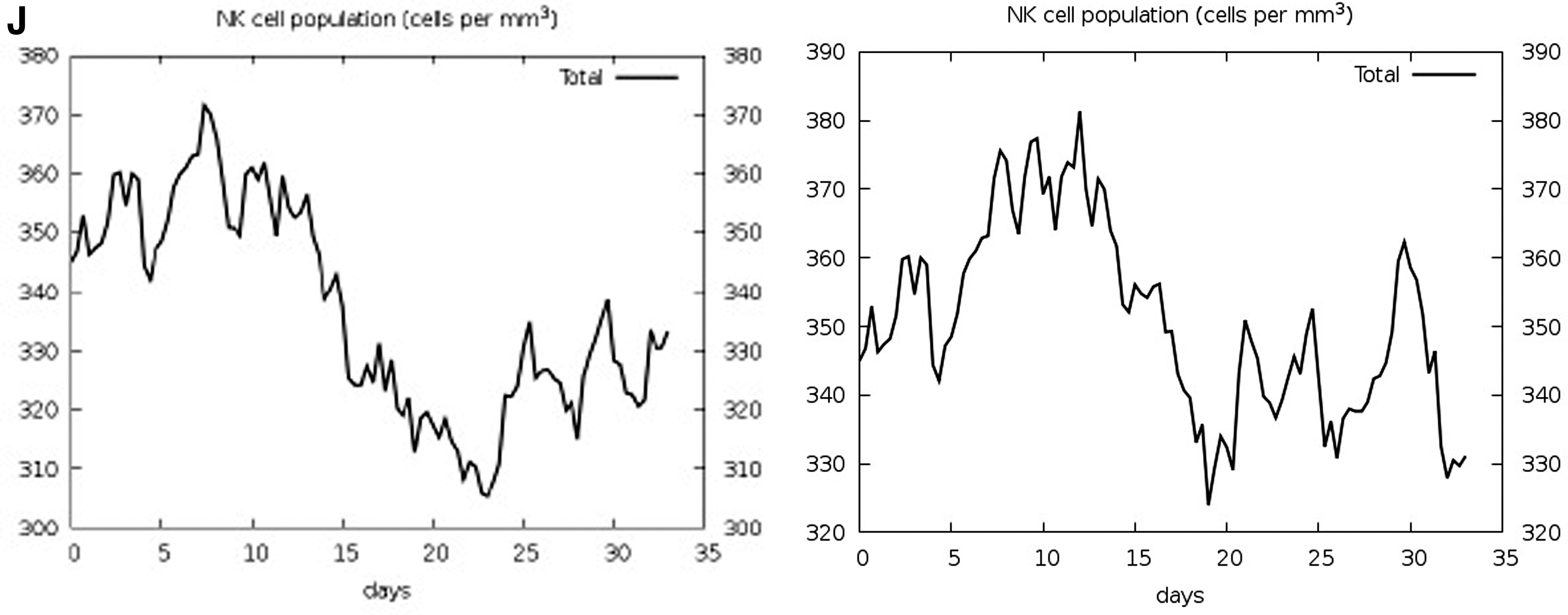
Figures 5G and H represent the immunoglobulin isotopes and the cytokines and interleukin (IL) profiles. High levels of IgM+IgG, IgG1+IgG2, and IgG1 were shown in the first 2 weeks upon administration of the vaccines. The induction of high levels of IFN-γ, IL-4, IL-12, tumor necrosis factor-β and -α, IL-10, and IL-6 indicates a good immune response. The levels of immunoglobulins and cytokines were relatively higher for Delta. High DC activity was observed during the construct exposure (Fig. 5I). A steady response of DC population per state (cells/mm3) was seen during vaccination with Delta. The increase and then decline in the proliferation of NK cells were shown (Fig. 5J).
Discussion
Despite the wide vaccination against SARS-CoV-2, the emergence of novel circulating mutants is a warning of a curb in the effectiveness of the vaccines and may need the annual revival of the virus vaccine like seasonal influenza vaccine. The vaccines target the S glycoprotein due to its ability to elicit strong immune responses within host cells. Mutations in the coding region of this gene lead to the generation of VOCs, which have been first recognized in the United Kingdom, Brazil, India, and South Africa then spread to other countries (Volz et al, 2021). At present, multiple SARS-CoV-2 variants that have the potential to cause more severe illness and evade either natural or vaccine-induced immune responses are circulating (Campbell et al, 2021).
Despite the effectiveness of the vaccines at an acceptable rate in controlled conditions, the last 2 years have witnessed several waves of SARS-CoV-2 that arise the question of whether the approved and or emergency authorized vaccines are effective against the new viral variants? In case of inefficiency, the presence of various variants threatens the global impact of mass vaccination campaigns and upsurges the concern to develop more effective vaccines or to change the vaccination strategy for alleviating the emergence of the virus variant. Comparing the genetic changes by focusing on their impacts on antigenicity of variants can help to understand the vaccine protection against the severe disease.
Considering the possibility of updating the vaccine sequence, we compared the characteristics of Delta and the Omicron in inducing immune responses in the mRNA vaccine platform as a perspective for further studies. An effective vaccine should contain both B cell and T cell epitopes to induce specific immune response against the immunogen. Results from B cell epitope prediction indicate slight differences in the positions and epitope sequences of S protein in Delta and Omicron, which did not alter the antigenicity of these variants. Based on the Kolaskar and Tongaonkar's algorithm, mutations in S protein of Omicron did not change its feature. The protein is potentially antigenic and can stimulate sufficient humoral response against SARS-CoV-2. The generation of a long-lasting immune response of T cells was evaluated by the highest binding affinity to the MHC molecules. No difference was observed in the prediction of CTL epitopes between the variants. Analysis in the IEDB MHC II binding prediction resulted in prediction of the lower amount of Median consensus percentile peptides in the Delta variant that reveal a stronger affinity to HTL alleles.
These findings were further confirmed by the molecular docking of the protein and human TLRs. Upon infection, SARS-CoV-2 binds to TLRs and generates an immune response following identification of the virus particles. Induction of the potent immune responses is highly related to the binding affinity of vaccine construct with the host TLRs (Mabrey et al, 2021; Patra et al, 2020). We selected TLR3, TLR4, and TLR7 which can recognize either the virus RNA genome or the S protein and docked them with the vaccine constructs to evaluate their capacity for interaction with immune receptors. The TLRs use both MyD88- and TRIF-dependent pathways to generate a type I/III IFN response and a robust inflammatory response through transcription factor nuclear factor kappa B (Choudhury et al, 2021). The docking energy scores and the smaller Kd revealed that the Delta variant has relatively higher binding interaction with all of the TLR3, TLR4, and TLR7 than Omicron, suggesting that the variant may better trigger TLR activation and facilitate to produce a strong immunity.
Differences in residue interaction of RBD protein with ACE2 (right panel of Fig. 3) represent the change in binding affinity of the Delta to ACE2 in comparison to the Omicron. Accordingly, the Omicron variant causes a milder infection than Delta if the ability depends mainly on the binding affinity. The strong binding free energies of RBD Delta to ACE2 have been attributed to the hydrophobic interaction of the residues at the 484–490 position. Eleven residues of Omicron variant are not involved in interaction with ACE2 resulting in a reduction in binding affinity to the human receptor and also the effectiveness of existing monoclonal antibodies. Therefore, the RBD of the new variant is under selective pressure to evade the host immune response and gain the ability to easy transmission among the vaccinated individuals. The high mutation frequency in RBD may significantly impact the translation efficiency of S protein and the generation of innate and adaptive immune responses against SARS-CoV-2. The high frequency of mutations in RBD may significantly impact the translation efficiency of the S protein-encoding region and thus the generation of innate and compatible immune responses against SARS-CoV-2 (Lin et al, 2022; Planas et al, 2022).
Based on the free energy level, the secondary mRNA structure of this region is more stable in the Delta variant than in the Omicron. More pseudoknots, especially bulge loop and interior loop, were observed in Omicron; however, insertion of these structures does not necessarily imply low translation efficiency.
Successful stimulation of the immune response and how to improve the half-life of the candidate vaccine were assessed. These constructs evoke the critical immune system components, including both B and T cell populations and memory cells. Although increased and stable levels of memory and active B cells respond to antigen response leading to a prolonged humoral response, the delay in IgG production in the first week indicates that the response alone is not sufficient for potent immunization. Therefore, an effective vaccine must elicit a cellular adaptive immune response. For both Delta and omicron, some of the predicted epitopes are overlapped and seem to have the ability to simultaneously induce cellular and humoral immunity. In particular, the production of IFN-γ was in line with the rapid response of NK cells (day 7). These cells are CTLs, and activation of them results in the production of IFN-γ, which regulates adaptive immunity (van Eeden et al, 2020). The higher frequency of matured DCs also affects T cell priming and activation, thereby better manipulating these immune responses (Langenkamp et al, 2000).
Interpretation of the simulation-based generated immune response revealed that both variant-specific vaccines are capable of generating an effective adaptive immune response against SARS-CoV-2; however, the Delta variant might be more effective than the new variant. The in silico results are consistent with serological data obtained from individuals vaccinated with the current mRNA vaccines. Antibody levels among the vaccinated people stay at higher levels compared to those vaccinated with inactivated vaccines and adenoviral vaccines if the early encoding sequence of the S gene is targeted to develop all of these vaccines (Chen et al, 2021; Xia et al, 2020; Yue et al, 2022). Serological data show a waning of antibody levels against Delta, which declined even further for Omicron. The vaccine's effectiveness against symptomatic disease caused by the new variant has been decreased by around 20% (Andrews et al, 2022; Campbell et al, 2021).
Because humoral immune responses may be less effective against newer strains of SARS-CoV-2, recent studies have emphasized the evaluation of protective immunity produced by antigen-specific T cells against this infection. It is estimated that the mRNA vaccine protection against hospitalization and death with Delta remains high (above 90% efficacy) (Andrews et al, 2022; Pouwels et al, 2021; Tartof et al, 2021). Although not enough information has been released about Omicron, it can be concluded that the current mRNA vaccines will protect against severe disease and death because T cells are unaffected by mutations in this variant.
In an ongoing pandemic, access to the appropriate vaccine in the early phase is critical to stemming the transmission of the pathogen. During the SARS-CoV-2 epidemic, most countries especially those with the lowest incomes received vaccines with delays that doubt the possibility of achieving herd or population immunity. In contrast, the emergence of new variants with increased transmissibility is changed in the herd immunity equation. An obstacle to the effective vaccine against SARS-CoV-2 is the differences in distribution and antigenicity of circulating VOCs from the selected vaccine strain resulting in a mismatch (Lazarevic et al, 2021; Rapaka et al, 2022). This situation is the greatest challenge for the global controlling campaign because it provides the possibility of immune escape mutations. In the long-term prospects, the SARS-CoV-2 pandemic will probably become an endemic disease like influenza. To improve vaccine effectiveness, seasonal influenza vaccines are updated based on the match between the selected vaccine viruses and those circulating (Hannoun, 2013).
Conclusion
Building infrastructure and processes to monitor the emergence of new variants of SARS-CoV-2 will enable the manufacturers to develop a safe and effective vaccine. Without considering possible setbacks, the timeline needed for vaccine manufacture based on a new variant and regulations for approval takes at least several months. Until then, it seems that the best strategy to reduce the impact of new variants on the effectiveness of current vaccines is to administer booster doses than formulate the vaccine based on each new virus variant.
Authors' Confirmation Statement
Dr. S.S. is from Razi Vaccine and Serum Research Institute (Karaj, Iran), and Dr. A.A.H. is from Bahyaar Sanaat Sepahan Company and the Isfahan University of Medical Sciences (Isfahan, Iran), all where education and research are the primary functions.
Footnotes
Authors' Contributions
Conception/design (equal), data collection (equal), data analysis and interpretation (S.S.), drafting the article (S.S.), review and editing (equal).
Data Availability Statement
The analyzed data are shared in this text.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study is co-supported by Razi Vaccine Serum Research Institute and Behyaar Sanaat Sepahan Company under grant number 13-18-1852-035-00025-000510.