
Abstract
This study established a new protocol of the antibody binding test to evaluate the potency of the rabies vaccine containing the final bulk and the product. The principle of this experiment is to combine rabies vaccine with quantitative anti-rabies virus neutralizing antibody. After combination, the remaining rabies vaccine is combined with the quantitative fluorescent labeled rabies virus. After this, observe the remaining fluorescent labeled rabies virus, calculate the fluorescence area with fluorescence observation equipment, then calculate the potency of rabies vaccine by Reed and Muench method. The test results of many batches of rabies vaccine final bulk and finished products showed that the potency detected by this method was consistent with that of National Institute of Health method.
Introduction
Rabies is a viral zoonotic disease responsible for an estimated 59,000 human deaths and >3.7 million disability-adjusted life years lost every year (Hampson et al, 2015). Rabies is almost invariably fatal once clinical signs appear, as a result of acute progressive encephalitis. Rabies occurs mainly in underserved populations, both rural and urban, and has been documented for >4,000 years (Tarantola, 2017). Vaccination and injection of rabies immunoglobulins are the only effective methods.
Although rabies vaccine has experienced different stages from the development of nerve tissue vaccines to purified cell culture and embryonated egg-based rabies vaccines and the product quality of rabies vaccine has been continuously improved (World Health Organization, 2018), the method of evaluating the potency of vaccine products has not broken through for many years. The National Institute of Health (NIH) method is the only potency detection method, this in vivo assay is an immunization challenge test, developed >60 years ago (Seligmann, 1973).
World Health Organization supports the concept of “3R strategy” (replacement, reduction, and refinement) of the use of in vivo methods for biologicals production and control (Milstien et al, 1996), and European recommendations encourage manufacturers and national control laboratories to implement the “3R strategy” of laboratory animal testing (EU, 2010).
Development, validation, and use of in vitro alternative approaches have now become a priority; they are not only ethically sound but can also reduce batch testing costs and shorten the time for results to hours instead of weeks (Stokes et al, 2012). So some other methods, such as enzyme-linked immunosorbent assay and antibody binding test (ABT), which are easier to carry out, are used as an auxiliary test for potency detection (Nedosekov et al, 2001). They are used by manufacturers to monitor the consistency of vaccine production.
In this study, the traditional ABT method (Barth et al, 1981) was improved by combining the practical operation techniques of rapid fluorescent focus inhibition test (RFFIT) (Krämer et al, 2009). The method was simple to operate, could well detect the potency of bulk and finished products, and could well evaluate the consistency of products between batches.
Materials and Methods
Cells, virus, and animal
BSR cells and CVS-11 (challenge virus standard) were all from the Institute of Zoonosis, Jilin University of China; The ICR mice were obtained from Liaoning Changsheng Biotechnology Co., Ltd. of China. Test department use license number is SYXK (JI) 2020-0005. The purchase and use of experimental animals fully comply with national regulations and guidelines.
Samples
A set of common test samples including rabies vaccine final bulk and products were provided by Changchun Zhuoyi Biological Co., Ltd. (Changchun, China).
Critical reagents
Dulbecco's modified Eagle medium (DMEM) and bovine serum albumin were bought from Thermo Fisher Scientific; DFA 5100 (rabies direct fluorescent antibody) was procured from Merck Millipore; SRV (National reference standard for potency test of rabies vaccine) (11.4 IU/mL) and HRIG (The sixth batch of Human Rabies Immunoglobulin national standard) (37.0 IU/mL) were provided by National Institutes for Food and Drug Control.
Equipment and materials
Biochemical incubator, 96-well plates, micropipettor, ImmunoSpot S6.
Methods
Serial dilutions of sample and SRV
For each independent assay, two independent dilution series were prepared using 50 μL of each test sample and 50 μL of SRV. No sample is plated in the blank control wells (columns 1 and 12). Then 100 μL of DMEM was distributed in all wells, 50 μL of each sample was distributed in row A, three dilution series were carried out, 50 μL was pipetted from row A to row B, this operation was repeated down to row H; in row H, the excess 50 μL was discarded from all the columns, at the end of the process, each well contains 100 μL of liquid.
Neutralization of SRV and samples with HRIG
The HRIG solution is diluted (1:200) to obtain the working solution at 0.2 IU/mL. Then 100 μL per well of HRIG working solution was dispensed in all the 96 wells of the microplate—including columns 1 and 12. The plates were placed in an incubator at +37°C ± 2°C for 1 h ± 5 min.
Neutralization of CVS-11 with remaining HRIG
CVS-11 challenge virus was diluted to obtain a working preparation with the final titer of 1 × 106 fluorescent focus unit/mL. Then 100 μL of freshly prepared working solution of CVS-11 was dispensed in each well of the plate—including columns 1 and 12. Then the solution was incubated at 37°C for 1 hour.
The remaining virus infects the cell
Cultured BSR cells were prepared in cell suspension at working concentration, 1 × 106/mL. Then 50 μL of freshly prepared working solution of BSR was dispensed in each well of the plate—including columns 1 and 12. The solution was incubated at 37°C ± 2°C for 1 h ± 5 min. The plate was placed in a 5% carbon dioxide incubator and cultured at 37°C ± 0.5°C for 24 h.
Acetone fixation
After incubation, the 96-well plate was taken out, the liquid was poured out, dried gently by patting, 50 μL of 80% acetone (precooling at −20°C) was added into each well, and it was placed in the cold storage at 2–8°C for 30 min (or −20°C for 6–8 min).
Fluorescence binding
Rabies direct fluorescent antibody (DFA) is a fluorescein isothiocyanate conjugated antibody against the rabies virus nucleocapsid protein. The rabies DFA solution was diluted (1:100) to obtain the working solution. Then 50 μL per well of rabies DFA working solution was dispensed in all the 96 wells of the microplate—including columns 1 and 12. The plates were placed in an incubator at +37°C ± 2°C for 1 h ± 5 min. Then it was washed twice with phosphate-buffered saline.
ImmunoSpot S6 evaluation
The fluorescent focus of virus-infected cells in each well was imaged using ImmunoSpot S6 equipment, and the percentages of fluorescent focus >50% and <50% were determined, so the sample titer was calculated using the Reed and Muench method.
Computational formula
lgED50 = lg(1/A) − (B–0.5/B–C) × lgN
Sample potency value (IU/mL) = 10(D–E) × S
A: Dilution <50% of the proportion of fluorescent focus
B: Percentage of fluorescent focus >50%
C: Percentage of fluorescent focus <50%
N: Dilution factor
D: lgED50 of sample
E: lgED50 of standard
S: Potency value of standard
Statistical analyses
Analysis of data was performed using Statistical Product and Service Solutions (SPSS) software (version 20.0, SPSS Inc., Chicago, IL). Two-tailed test was applied for statistical analysis with alpha level set at α = 0.05. A p-value of <0.05 (p < 0.05) was considered statistically significant.
Results
This improved ABT can clearly show the virus fluorescent focus and the percentage of virus fluorescent focus in each hole, as shown in Figure 1. In this way, we can get relatively accurate potency value.
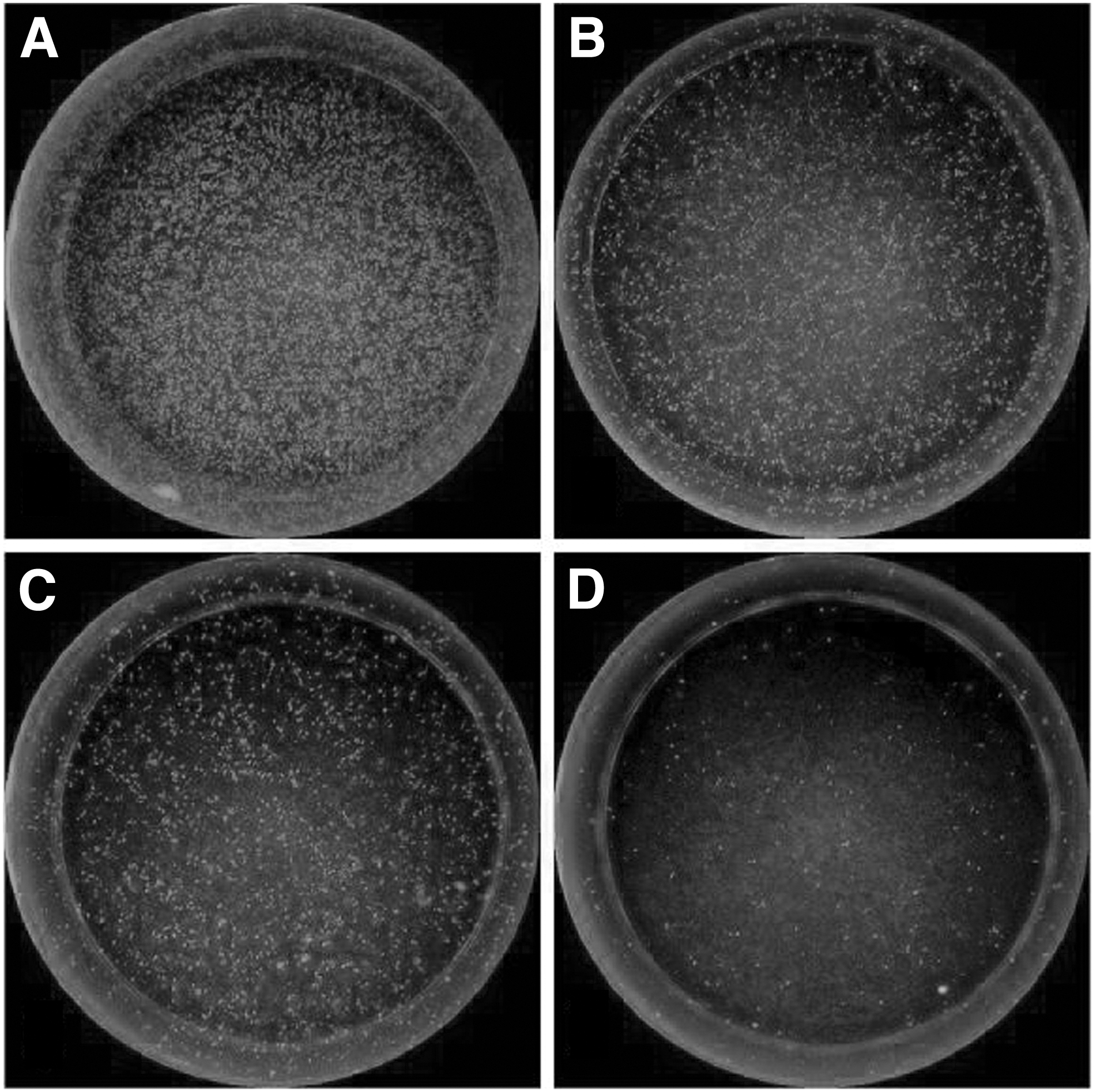
Immunofluorescence focus of virus-infected cells.
Repeatability of the improved ABT
Assay as shown in Table 1, three batches of final bulk and three batches of finished product samples were tested by the improved ABT method. The results showed that the coefficient of variation of the method was 2.4–5.7%, indicating that the repeatability of the method was good.
Repeatability of Our Improved Antibody Binding Test
CV, coefficient of variation; SD, standard deviation.
Comparison with NIH
The titers of 20 batches of rabies vaccine products were determined by the improved ABT method, and the results were compared with those of NIH mouse assay. The results of the two assay methods were similar, both >4 IU/mL, and the results of the improved ABT assay were more stable than those of NIH assay, as shown in Figure 2.
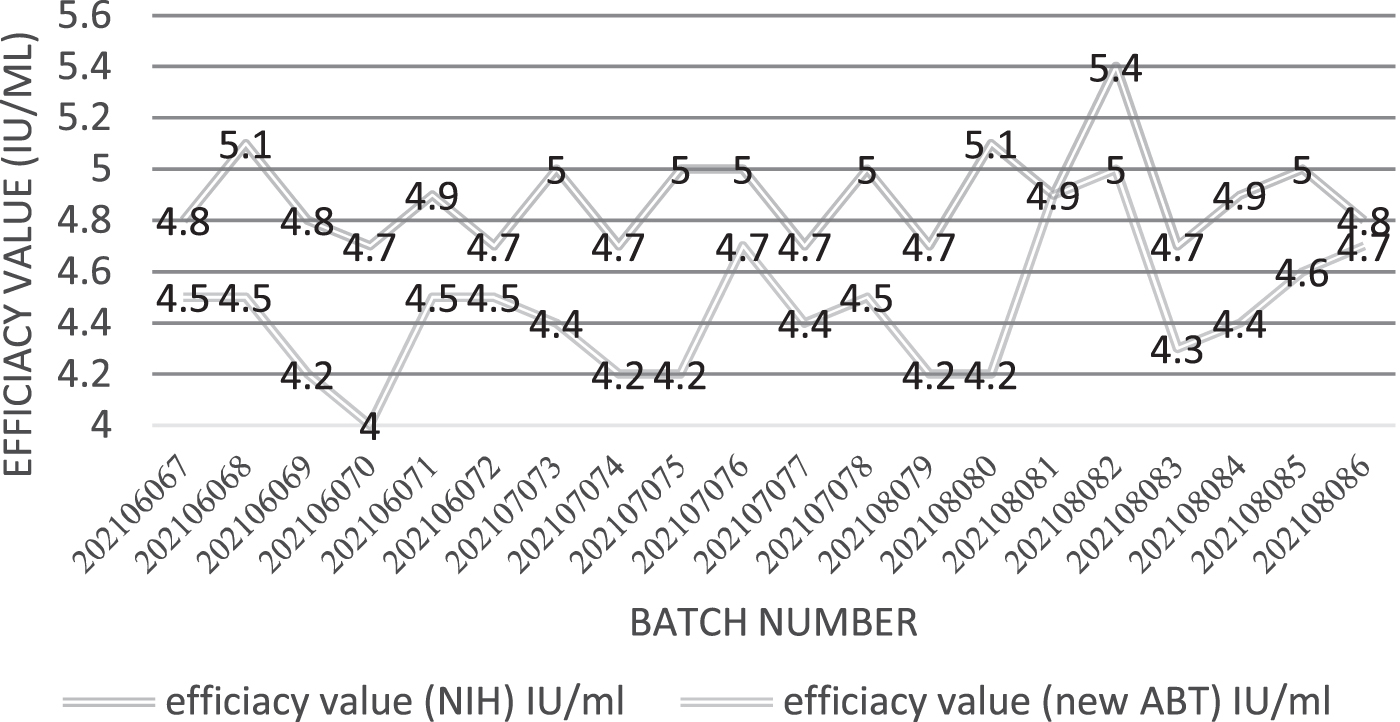
The potency value of 20 batches rabies vaccine results uses the NIH method and new ABT method. ABT, antibody binding test; NIH, National Institute of Health.
Twenty vaccine samples were tested by the new ABT method and the potency of each sample was re-evaluated using the NIH test. As shown in Figure 3, the results showed that the correlation coefficient of the two methods is 0.524, and the Significant value is 0.018, <0.05, indicating that the correlation between the two methods is significant.
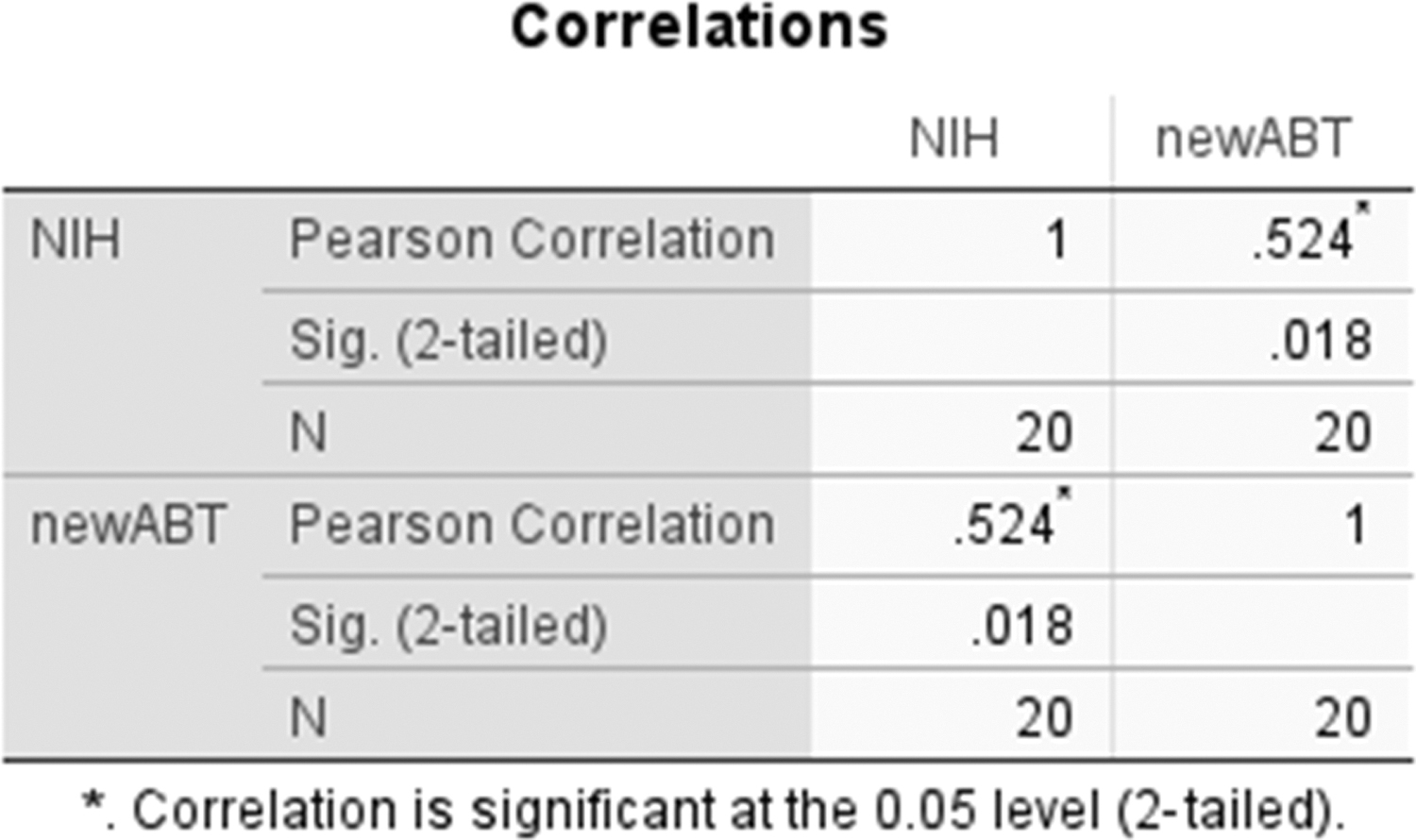
Comparisons of the new ABT and NIH test results for evaluation of the potency of rabies vaccine products.
Discussion
This new ABT method simplifies sample dilution steps, all dilutions can be performed in a 96-well plate. Moreover, it is more accurate to dilute with micropipe over the traditional ABT method, which requires the use of a separate test tube for sample dilution (Barth et al, 1981).
In addition, this method uses a fluorescence range reading device, which can take pictures of each plate hole in the 96-hole plate clearly. The fluorescence area ratio of each plate hole can be calculated through software, thus making the judgment and calculation of the results more accurate, pictures as Figure 1.
This article explains the calculation formula in detail, so that researchers who read this paper can accurately use the formula when establishing this method.
There are three key points in this method. The first is the working concentration of HRIG, which is determined by the sample. The working concentration of HRIG can just offset the sample with the highest concentration, which will avoid the failure of the test due to the excessive use of HRIG. The second is to determine the working concentration of CVS-11 standard attack virus. The standard attack virus infects cells so that the percentage of fluorescence foci in each cell well is 80% concentration, which is the optimal working concentration.
The determination of these two concentrations is key steps in the successful establishment of the method. Third, rabies DFA is an antibody to rabies virus nucleoprotein. Koraka et al (2014) confirmed that the glycoprotein trimer of rabies virus can well induce the body to produce neutralizing antibodies, thus playing a protective role. However, the effect of glycoprotein monomers on inducing neutralizing antibodies is poor (Koraka et al, 2014). The detection antibody used in this method is the fluorescent antibody of rabies virus nucleoprotein, which avoids the phenomenon that glycoprotein monomer can be detected by using glycoprotein monoclonal antibody, and the titer detection result is higher than the real effective value.
The repeatability test results show that the method repeatability is very good, the coefficient of variation of the maximum value is 5.7%. The biggest advantage of this method is that we can directly see the proportion of fluorescent focus through the microscope. The percentage value is calculated using microscope software, the result is more objective.
The principle of this method is that the effective components in the sample compete with CVS-11 to bind a certain amount of HRIG. The higher the percentage of fluorescent focus was observed, the higher the content of effective component of rabies vaccine. Therefore, if we observe that the percentage of immunofluorescence focus of the finished product is higher than a certain value of the standard, the potency of the product must be higher than that of the standard used for detection. This method tests 20 batches of finished products, there is no difference with the NIH method, as shown in Figure 2. This method can be used to quickly determine whether the product produced by the vaccine company is qualified or not. Make the product quality under effective control.
Conclusions
In this article, a new ABT method was established, which was simple to use, objective, and accurate to determine the fluorescence percentage, and the calculation formula was disclosed. Meanwhile, the repeatability of the method was verified, the results of the NIH method for 20 production batches of vaccines were compared. The results showed that the repeatability and comparability of the method with those of the NIH method were very good. Therefore, this article provides an alternative quality control method for rabies vaccine researchers and manufacturers. Through the description of this article, readers can easily establish this method, which is conducive to the communication between rabies vaccine researchers and method improvement.
Footnotes
Author Disclosure Statement
The authors declare no competing financial interests.
Funding Information
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.