
Abstract
Hepatitis B virus (HBV) infection is a major health problem worldwide and causes almost one million deaths annually. The HBV core gene codes for two related antigens, known as core antigen (HBcAg) and e-antigen (HBeAg), sharing 149 residues but having different amino- and carboxy-terminals. HBeAg is a soluble variant of HBcAg and a clinical marker for determining the disease severity and patients' screening. Currently available HBeAg assays have a shortcoming of showing cross-reactivity with HBcAg. In this study, for the first time, we evaluated whether HBcAg-adsorbed anti-HBe polyclonal antibodies could specifically recognize HBeAg or still show cross-reactivity with HBcAg. Recombinant HBeAg was cloned in pCold1 vector and successfully expressed in Escherichia coli and after purification by Ni-NTA resin was used to generate polyclonal anti-HBe antibodies in rabbit. Purified HBeAg was further characterized by assessing its reactivity with anti-HBe in the sera of chronically infected patients and HBeAg-immunized rabbit. Sera from patients with chronic HBV infection, containing anti-HBe, specifically reacted with recombinant HBeAg, implying antigenic similarity between the prokaryotic and native HBeAg in the serum of HBV-infected patients. In addition, the designed enzyme-linked immunosorbent assay (ELISA) with rabbit anti-HBe polyclonal antibodies could detect recombinant HBeAg with high sensitivity, while high cross-reactivity with HBcAg was observed. It is noteworthy that HBcAg-adsorbed anti-HBe polyclonal antibodies still showed high cross-reactivity with HBcAg, implying that due to the presence of highly similar epitopes in both antigens, HBcAg-adsorbed polyclonal antibodies cannot differentiate between the two antigens.
Introduction
More than half of the world's population has been exposed to the hepatitis B virus (HBV). As one of the leading causes of death among infectious disease, HBV infection is a major health problem worldwide (WHO, 2018). It is estimated that there are more than 245 million chronic carriers worldwide and almost a third of them will develop severe HBV-related complications, such as cirrhosis and hepatocellular carcinoma (Ganem and Prince, 2004; Lim et al., 2020; MacLachlan and Cowie, 2015).
HBV genome encodes four overlapping open reading frames (ORFs) (Lamontagne et al., 2016). Precore/core ORF encodes two distinct proteins: the core protein, which forms building blocks of nucleocapsid particles (HBcAg), and the secreted nonparticulate precore protein (HBeAg) (Eren et al., 2018). Extra 29 N-terminal amino acids of precore protein serve as a signal peptide and direct the nascent polypeptide to the secretory pathway. The 19-amino acid of signal peptide is cleaved during transferring into the endoplasmic reticulum, producing an adult precore protein with a molecular weight of 22 kDa (p22), which still contains 10 amino acids at the N-terminal (the propeptide: SKLCLGWLWG) (Garcia et al., 1988; Standring et al., 1988).
Therefore, the remaining p22 is identical to HBcAg except that it contains 10-residue propeptide. The remaining polypeptide is C-terminally truncated variably between residues 149 and 154 (at arginine-rich region), which is due to the heterogeneity in C-terminal cleavage sites, and eventually generates multiple species of mature HBeAg of 15–18 kb (Baumeister et al., 2000; Ou, 1997).
Although HBeAg and HBcAg share very similar primary structures, identical 1–149 amino acids, they differ in 10 amino acids at the N-terminal and 24–29 amino acids at the C-terminal (Baumeister et al., 2000; Wang et al., 2021).
HBcAg and HBeAg have several cysteine residues, but only cysteines −7 and 61 play a crucial role in their structure and in the eventual formation of either capsid particles or secretory variants (Watts et al., 2011). The cysteines at positions −7 of the propeptide and 61 are crucial for formation of an intramolecular S-S bond in HBeAg and secretion of this antigen and not formation of the capsid; presence of the cysteine −7 blocks the formation of any intermolecular disulfide bonds (DiMattia et al., 2013; Nassal and Rieger, 1993). However, in HBcAg, due to the absence of cysteine −7, an intermolecular disulfide bond between Cys-61-Cys-61 forms to bridge two neighboring monomers (Wynne et al., 1999). Therefore, the differences in the structure and physiology of HBeAg and HBcAg could be attributed to the presence of propeptide and its cysteine residue (Watts et al., 2011).
Full-length HBcAg has 183 amino acids and comprised two domains: an assembly domain spanning amino acids 1–149, and an arginine-rich nucleic acid-binding domain spanning amino acids 150–183 (Packianathan et al., 2010; Wynne et al., 1999). Both full-length HBcAg (1–183) and assembly domain of HBcAg (1–149) can form capsid particles (Zhuang et al., 2017; Zlotnick et al., 1997). As a building block of the virus nucleocapsid, HBcAg plays an essential role in the HBV replication cycle. On the contrary, the biological role of HBeAg, as a secreted antigen in unassembled form, is to establish immune tolerance, modulate the host immune response, and eventually developing chronic infection (Eren et al., 2018; Kramvis et al., 2018).
The level of serum HBeAg is associated with the virus replication and active hepatitis B infection. HBeAg seroconversion is a sign of successful therapy in chronically infected patients. Therefore, quantification of HBeAg is a clinically useful marker for determining the status of the infection and could be monitored to predict the response to treatment (Bernard et al., 1997; Kimura et al., 2002). One of the major shortcomings of available HBeAg assays is cross-reactivity with HBcAg. Therefore, developing sensitive assays with specific anti-HBe antibodies that show no cross-reactivity with HBcAg seems necessary (Wang et al., 2021; Zhuang et al., 2017). Although HBeAg and HBcAg show high structural similarity, several anti-HBe monoclonal antibodies that differentially recognize HBeAg and show no reactivity to HBcAg have been generated so far (Sogut et al., 2011; Wang et al., 2021; Zhuang et al., 2017). However, the specific reactivity of HBcAg-adsorbed anti-HBe polyclonal antibodies with HBeAg has not been assessed so far.
In this study, His-tagged HBeAg (amino acids −10 to 149) was cloned in Escherichia coli and, after purification, its reactivity with anti-HBe in the serum of HBV-infected patients was assessed to determine whether prokaryotic HBeAg has similar antigenicity to native HBeAg. Next, prokaryotic HBeAg was used to generate rabbit polyclonal antibodies, which could be eventually used for the development of sensitive HBeAg enzyme-linked immunosorbent assay (ELISA). In addition, to obtain anti-HBe polyclonal antibodies that specifically react with HBeAg and not HBcAg, we assessed whether adsorption of anti-HBe polyclonal antibodies with HBcAg coupled to the Sepharose column (HBcAg-adsorbed anti-HBe antibody) could result in HBeAg-specific polyclonal antibodies.
Materials and Methods
Materials and bacterial strains
The following reagents and kits were used in this study: plasmid extraction kit, gel purification kit, T4 DNA ligase, restriction enzymes, Taq DNA polymerase, nitrocellulose Western blotting membranes, mouse anti-human IgG-HRP, Freund's complete and incomplete adjuvants (Sigma-Aldrich, Germany), HiTrap Protein G HP (GE Healthcare, United Kingdom), tetramethylbenzidine (TMB; Pishtazteb, Iran), and strains of bacteria (Pasteur Institute, Iran). Anti-HBeAg-positive sera were collected from the Gholhak Clinical Laboratory, Iran.
Construction of recombinant HBeAg
The DNA sequence corresponding to HBeAg (amino acids −10 to 149) was amplified from plasmid pT-HBV1.3 (kindly provided by professor Protzer) by polymerase chain reaction (PCR) using forward (NdeI-22b-HBeAg-F: 5′-GTTTTTTCATATGTCCAAGCTGTGCCTTG-3′) and reverse (HindIII-22b-HBeAg-R: 5′-GGTTGTGAAGCTTAACAACAGTAGTCTCC-3′) primers that contained NdeI and HindIII restriction sites, respectively.
The amplified product was digested by appropriate restriction enzymes (Thermo Scientific) and then inserted downstream of the hexa-histidine tag sequence between NdeI and HindIII sites of pColdI vector (Takara, Japan). Therefore, the recombinant vector encoded six histidine residues at N-termianl of HBeAg. The recombinant pColdI plasmid containing HBeAg (HBeAg-pColdI) was sequenced to verify the orientation and sequence of HBeAg.
Recombinant HBeAg expression in E. coli
The recombinant plasmid HBeAg-pColdI was simultaneously transformed and expressed in six commercialized E. coli expression hosts, including BL21 (DE3), Rosetta-gami (DE3) plysS, Origami (DE3), Novablue (DE3), Tuner (DE3), and Shuffle (DE3), and their ability for expression of recombinant HBeAg was compared. Different HBeAg-pColdI transformed hosts were grown at 37°C in 5 mL of Luria Broth (LB) medium supplemented with 100 μg/mL of ampicillin in a shaker incubator (250 rpm) for 16 h. Next, 600 μL of the overnight culture was used to inoculate 10 mL of LB media containing 100 μg/mL of ampicillin and incubated at 37°C in a shaker incubator (250 rpm) until the optical density (OD) at 600 nm was between 0.9 and 1. At this time, isopropyl-Dthiogalactopyranoside (IPTG) (Sinaclone, Iran) was added to the culture to a final concentration of 1 mM and incubated at 15°C in a shaker incubator for 24 h. Samples were collected at different time points (0, 2, 4, 6, 24 h) postinduction to determine the maximum yield.
Samples were resuspended in 2 × sample loading buffer and the proteins were separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels and detected by Coomassie Brilliant Blue staining.
Purification of recombinant HBeAg
Transformed E. coli stock cells were inoculated into 10 mL of LB broth containing 100 μg/mL of ampicillin and allowed to grow overnight in a shaking incubator at 250 rpm, 37°C. This culture was used for inoculation of 250 mL of LB broth containing 100 μg/mL of ampicillin and then the culture was grown in a shaking incubator at 250 rpm, 37°C until OD600 reached 0.9–1. At this point, the culture was induced with 1 mM of IPTG and incubated at 15°C and allowed to grow in a shaking incubator at 250 rpm for 24 h, and then cells were pelleted by centrifugation at 4,000 rpm (2,665 g) for 20 min at 4°C. Harvested bacterial pellets containing inclusion bodies were solubilized in 7.5 mL of lysis buffer1 (100 mM NaCl, 100 mM NaH2PO4, 30 mMTris-HCl, pH = 7.8) and incubated on ice for 40 min.
This solution was continuously sonicated (Hielscher UP200H, Germany) on ice for 12 cycles of 20 sec at 90% amplitude with 40 sec of cooling in between the cycles to lyse the cells. The lysate was then centrifuged at 12,850 g for 20 min at 4°C to separate the soluble and insoluble fractions. The purified inclusion bodies' pellet was dissolved in 7.5 mL of lysis buffer 2 (300 mM NaCl, 100 mMNaH2PO4, 10 mMTris-HCl, 10 mM imidazole, 6 M urea, pH = 7.8) and after incubation on ice for 30 min, the suspension was centrifuged at 16,000 g for 20 min at 4°C. The pellet was discarded and the supernatant containing His-tagged recombinant HBeAg was incubated with Ni-NTA agarose (Qiagen, Germany) beads, which were already equilibrated with lysis buffer 2, for 3–4 h at room temperature (RT) on an end-over-end shaker.
Following the binding of recombinant HBeAg, the resin was washed with lysis buffer 2 and then, elution was started by applying a wash buffer (300 mM NaCl, 100 mM NaH2PO4, 40 mM imidazole, 6 M urea, pH = 8) to detach nonspecific proteins from the resin. Then, recombinant HBeAg was eluted by an elution buffer (500 mM NaCl, 100 mM NaHCO3, 500 mM imidazole, 2 M urea, pH = 8.3).
The purity of the fractions containing purified HBeAg was determined by SDS-PAGE and the fractions were dialyzed against a dialysis buffer (500 mM NaCl, 100 mM NaHCo3, 2 M urea, pH = 8.3).
Evaluation of antigenic structure of HBeAg
Antigenic similarity between the recombinant and native HBeAg was evaluated with three assays. Reactivity of recombinant HBeAg was evaluated with the serum of patients containing anti-HBe in both indirect ELISA and Western blot. In addition, a commercial HBeAg/HBeAb detection kit (Diapro, Italy) was used to check whether it can recognize our recombinant HBeAg.
Western blotting analysis
One microgram per well of recombinant HBeAg in reduced and nonreduced conditions was loaded on polyacrylamide gel, and then transferred to a nitrocellulose membrane using an electroblot system (BioRad, Hercules, CA) at 110 V for 75 min. The membrane was blocked in 5% skim milk, at 4°C for 16 h. After three times washing with phosphate-buffered saline (PBS) containing 0.05% Tween 20 (PBST), the membrane was incubated with patients' sera (eight pooled sera obtained from Gholhak Pathobiology Laboratory, Tehran) containing a high titer anti-HBe antibody at 1/1,000 dilution. After incubation for 2 h at RT, the membrane was washed three times and an appropriate concentration of HRP-conjugated mouse anti-human IgG was added and incubated for 1 h at RT. After washing, the membrane was detected using the ECL Prime Western Blotting Detection Reagent, (Thermo Scientific).
Indirect ELISA
Reactivity of recombinant HBeAg was evaluated with the sera of patients containing anti-HBe in an indirect ELISA. Briefly, plates were coated with different concentrations of recombinant HBeAg (10, 5, 2.5, 1.25, and 0.62 μg/mL) at 4°C for 16 h. Plates were then washed by PBST and blocked with 3% skim milk for 1 h at 37°C. Patients' sera containing high titer anti-HBe (pooled sera obtained from Gholhak Clinical Laboratory) at 1/1,000 dilution and also normal sera were added and incubated for 1 h at 37°C. Next, HRP-conjugated mouse anti-human was added and incubated for 1 h at 37°C. After addition of TMB, OD was measured at 450 nm by a multiscan ELISA reader (BioTek, Winooski).
Reactivity of recombinant HBeAg with commercial ELISA kit
Reactivity of our recombinant HBeAg was assessed with a commercial ELISA kit (Diapro, Italy) using mouse monoclonal anti-HBe as capture and detection antibody, according to the manufacturer's instruction.
Production, purification, and characterization of anti-HBe polyclonal antibody
Two female New Zealand white rabbits (Razi Vaccine and Serum Research Institute, Karaj, Iran) were immunized subcutaneously with 50 μg of recombinant HBeAg dissolved in 500 μL of PBS and emulsified in 500 μL of complete Freund's adjuvant. Four weeks later, the animals were immunized with 25 μg of recombinant HBeAg in incomplete Freund's adjuvant every 2 weeks until the anti-HBe antibody titer reached a plateau. Titer of anti-HBe polyclonal antibody was assessed by ELISA. Then, to obtain HBeAg-purified anti-HBe polyclonal antibodies, animal sera were purified by the immunoaffinity column prepared in our laboratory by coupling 2 mg of purified HBeAg to 1 g of CN-Br-activated Sepharose4B (GE Healthcare) according to the manufacturer's instruction. It is noteworthy that HBeAg used for immunization of the animals was in a buffer containing 500 mM NaCl, 100 mM NaHCo3, 2 M urea, pH = 8.3.
To evaluate the specific reactivity of HBeAg-purified anti-HBe polyclonal antibodies with HBeAg, a sandwich ELISA was designed. Ten micrograms per milliliter of HBeAg-purified anti-HBe polyclonal antibody was coated on an ELISA plate and incubated at 4°C for 16 h. After washing, the wells were blocked with 3% BSA prepared in 0.05% PBST. Next, different concentrations of recombinant HBeAg (20, 10, 2.5 ng/mL) and also native HBeAg were added and incubated at 37°C for 1 h. After washing, biotin-conjugated HBeAg-purified anti-HBe was added. 1 IU/mL antigen calibrator of HBeAg commercial kit (Diapro) was used as positive control. After washing and adding streptavidin-HRP, TMB was added and ODs were detected.
Two sources of native HBeAg were used in the experiment. In this study, HepG2-NTCP cells were infected with HBV inoculum prepared after concentrating culture supernatant of HepAD38 cell line (Asadi-Asadabad et al., 2021). Five, 9, and 14 days after infection, the supernatant containing native HBeAg was collected and used in this experiment. In addition, serum of patients infected with HBV containing native HBeAg was included in this experiment. The presence of HBeAg in the serum of HBV-infected patients and the supernatant of HBV-infected HepG2-NTCP cells were already confirmed by the commercial HBeAg ELISA kit (Diapro).
To evaluate the reactivity of anti-HBe polyclonal antibodies with sequential and conformational epitopes, we also performed an ELISA with reduced and nonreduced HBeAg. Different concentrations of HBeAg (0.2–2.0 μg/mL) were incubated with 2-mercaptoethanol (final concentration 20 mM) at 37°C for 1 h and then coated on an ELISA plate and incubated at 37°C for 1.5 h. After blocking with 3% BSA, biotin-conjugated HBeAg-purified anti-HBe was added followed by streptavidin-HRP. Finally, TMB was added and ODs were detected.
In all experiments, animals were housed and handled according to the guidelines of the Ethics Committee of Tehran University of Medical Sciences, in accordance with the principles of the Helsinki Declaration.
Production and assessing the specific reactivity of HBcAg-adsorbed anti-HBe polyclonal antibody
Although HBeAg and HBcAg show a high structural similarity, several anti-HBe monoclonal antibodies that differentially recognize HBeAg and show no reactivity to HBcAg have been generated so far (Wang et al., 2021; Zhuang et al., 2017). Therefore, we decided to pass HBeAg-purified anti-HBe polyclonal antibodies through the HBcAg-CNBr Sepharose column to obtain HBcAg-adsorbed-anti-HBe polyclonal antibodies and assess whether these polyclonal antibodies can specifically and differentially recognize HBeAg or still show cross-reactivity with HBcAg. Accordingly, recombinant HBcAg was generated with a similar strategy used for generating recombinant HBeAg. Briefly, the HBcAg sequence (amino acids 1 to 149) was amplified from plasmid pT-HBV1.3 by PCR using forward (NdeI-22b-HBcAg-F: 5′-GGTTTGTCATATGATGGACATCGACCCTTATAA-3′) and reverse (HindIII-22b-HBeAg-R: 5′-GGTTGTGAAGCTTAACAACAGTAGTCTCC-3′) primers that contained NdeI and HindIII restriction sites, respectively.
The PCR, expression, and purification were carried out with similar conditions with HBeAg. The HBcAg-CNBr Sepharose immunoaffinity column was prepared in our laboratory by coupling 2 mg of purified HBcAg to 1 g of CN-Br-activated Sepharose4B (GE Healthcare) according to the manufacturer's instruction.
To evaluate whether HBcAg-adsorbed anti-HBe polyclonal antibody can differentially recognize HBeAg and not HBcAg, the specific reactivity of the HBcAg-adsorbed antibodies was analyzed by ELISA (sandwich and indirect) and Western blotting.
For sandwich ELISA, 10 μg/mL of HBcAg-adsorbed anti-HBe polyclonal antibody was coated on the ELISA plate and incubated at 4°C for 16 h. After blocking with 2%BSA, different concentrations of HBcAg and HBeAg were added separately. Then, biotin-conjugated HBeAg-purified anti-HBe at a dilution of 1/4,000 was added. Finally, streptavidin-HRP was added and after adding TMB, the ODs were recorded.
For indirect ELISA, 0.5 μg/mL of HBeAg and HBcAg was coated on the ELISA plate and incubated at 4°C for 16 h. After blocking with 3% skim milk, different concentrations of HBeAg-purified anti-HBe polyclonal antibody, HBcAg-adsorbed anti-HBe polyclonal antibody, and also HBeAg-bound anti-HBe polyclonal antibody at different concentrations of 4, 1, 0.25, and 0.06 μg/mL were added and incubated at 37°C for 1 h. After washing, sheep anti-rabbit-HRP (Sigma-Aldrich) at a concentration of 1/2,000 was added. Finally, after adding TMB, the ODs were recorded.
For Western blotting, 0.5 μg/mL of HBeAg and HBcAg in nonreduced condition was loaded on 13% polyacrylamide gel, and then transferred to the nitrocellulose membrane. The membranes were blocked with 5% skim milk at 4°C for 16 h. After washing, the membranes were incubated with 1 μg/mL of HBeAg-purified anti-HBe polyclonal antibody and HBcAg-adsorbed anti-HBe polyclonal antibody. After incubation for 1 h at RT, the membrane was washed and an appropriate concentration of HRP-conjugated sheep anti-rabbit was added and incubated for 1 h at RT. After washing, the membrane was detected using the ECL Prime Western Blotting Detection Reagent.
Results
Molecular cloning, expression, and purification of recombinant HBeAg
The HBeAg gene (Fig. 1) (containing amino acids −10 to 149) was amplified and inserted into expression vector pColdI between the Nde1 and HindIII restrictions sites (Fig. 2). After sequencing and verification of HBeAg gene sequence, the recombinant plasmid HBeAg-pColdI was transformed and expressed in E. coli BL21 strain.

The sequence of HBeAg (−10)−149 used in this study. The first and last 17 and 8 residues (underlined), respectively, were derived from pCold1 vector. The 10 bold amino acids represent residues corresponding to 10 precore amino acids (−10 to −1).

Schematic representation of restriction map and multiple cloning site (MCS) of pColdI vector. Arrows show the position of the restriction sites used for insertion of HBeAg in the vector.
SDS-PAGE analysis of the total cell lysate showed that bacterial culture induced with 1 mM IPTG showed overexpression of HBeAg band, while in the before induction sample, no overexpression was observed. As shown in Figure 3A, a clear band of overexpression was observed with a molecular mass of almost 21 kDa, containing a His-tag at the N-terminal of HBeAg. It is noteworthy that the majority of the expressed protein was in the insoluble (inclusion body) fraction of cells. Among six different E. coli expression hosts, including BL21, Rosetta-gami, Origami, Novablue, Tuner, and Shuffle, the highest expression level of recombinant HBeAg was obtained in Rosetta-gami strain (Fig. 3B).

SDS-PAGE analysis of HBeAg expression.
Recombinant HBeAg containing his-tag at its N-terminal was purified by NiNTA resin. It is noteworthy that the presence of the majority of this protein in the inclusion body appeared to be due to its low solubility. In addition, elution of HBeAg in a buffer with no urea resulted in the accumulation of insoluble aggregates, which was avoided by keeping 2 M urea in the elution buffer.
SDS-PAGE analysis of purified fractions (lines 6–10) showed a highly pure preparation of HBeAg. However, some faint bands, which might be the impurities, are seen below and above the mail 17 Kd band.
The yield was calculated from the total amount of purified HBeAg in the final volume of LB media, which was ∼4 mg/L (Fig. 4).
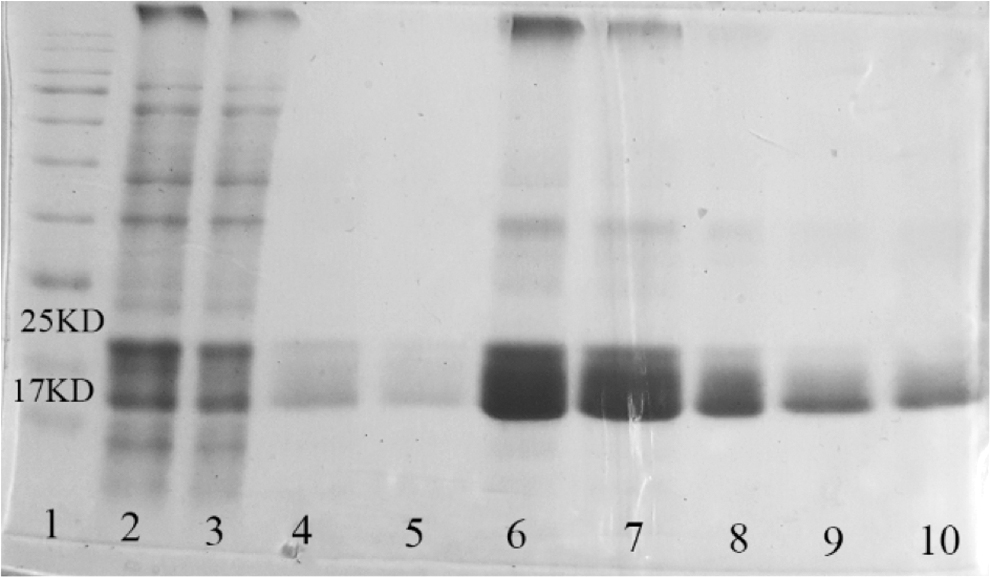
SDS-PAGE analysis of HBeAg purification by Ni-NTA affinity chromatography. Lane1: protein ladder, lane 2: lysate of HBeAg-pColdI transformed bacteria after induction with IPTG, lane3: flow-through fraction (unbound proteins), lanes 4 and 5: wash fractions, lanes 6–10: elution fractions containing recombinant his-tagged HBeAg. IPTG, isopropyl-Dthiogalactopyranoside.
Assessing the antigenic structure of HBeAg
Anti-HBe antibodies in the serum of chronically infected patients can recognize recombinant prokaryotic HBeAg in Western blot
To confirm the structural similarity of the purified recombinant HBeAg to the native one, reactivity of the anti-HBe antibodies in a pooled sera of chronically infected patients with recombinant HBeAg was checked in Western blot and ELISA. As Figure 5 shows, HBeAg in forms of monomer, dimer, and trimer with approximate sizes of 17, 35, and 70 kDa, respectively, was detected by anti-HBe antibodies generated against native HBe in patients' sera. It is noteworthy that anti-HBe antibodies in the pooled sera were also able to recognize HBeAg in a reduced condition. Due to the intramolecular disulfide bond in rHBeAg, the protein in the nonreduced condition has slightly higher mobility than the reduced condition (Zhuang et al., 2017).

Specific reactivity of anti-HBe antibodies in sera of HBV-infected patients with recombinant HBeAg in Western blot. Lane 1: lysate of HBeAg-pColdI-transformed bacteria after induction with IPTG in nonreduced (lane 1) and reduced (lane 6) conditions treated with pooled sera of HBV-infected patients, lanes 2 and 3: 1 and 0.5 μg of purified recombinant HBeAg, respectively, in nonreduced condition, lane 4: 1 μg of purified recombinant HBeAg in reduced condition treated with pooled sera of HBV-infected patients, lane 5: lysate of HBeAg-pColdI transformed bacteria after induction with IPTG treated with serum of normal subject. HBV, hepatitis B virus.
Anti-HBe antibodies in the serum of chronically infected patients can recognize recombinant prokaryotic HBeAg in ELISA
To assess the antigenicity of recombinant prokaryotic HBeAg, specific reactivity of anti-HBe antibodies in the serum of HBV-infected patients with recombinant HBeAg was also evaluated in ELISA. As Figure 6 shows, anti-HBe antibodies, elicited against native HBeAg in the serum of HBV-infected patients, recognized recombinant HBeAg at different concentrations (10 to 0.62 μg/mL), while serum of normal subject showed no reactivity as negative control.
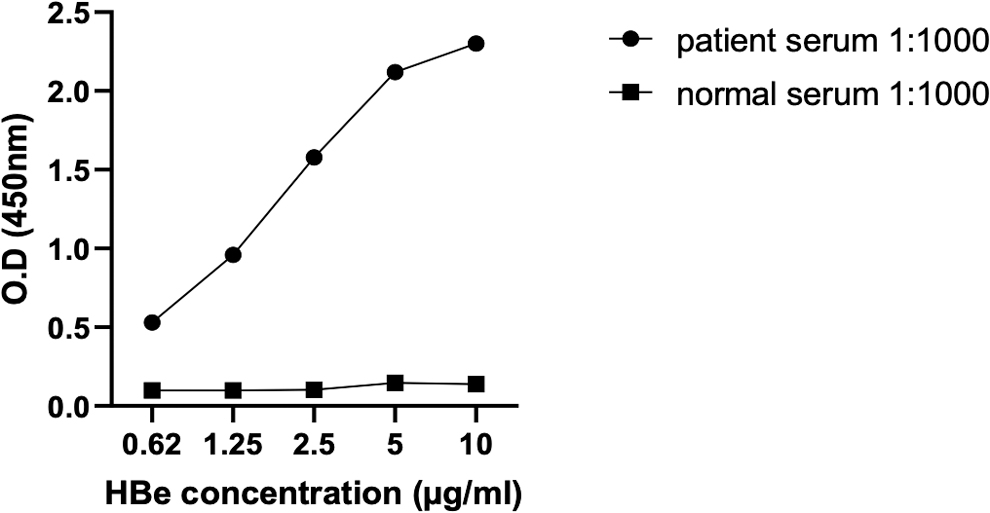
Specific reactivity of anti-HBe antibodies in the sera of HBV-infected patients with recombinant HBeAg in ELISA. ELISA plate was coated with different concentrations of recombinant HBeAg and 1/1,000 dilution of pooled sera of HBV-infected patients and also normal subject, as negative control, was used. ELISA, enzyme-linked immunosorbent assay.
Reactivity of recombinant HBeAg with commercial HBeAg/Ab detection kit
To confirm that the structure of purified recombinant HBeAg is similar to that of native HBeAg, a commercial HBeAg/Ab detection kit (Diapro, Italy), commonly used in our country, was used. As Figure 7 shows, the commercial kit was able to detect purified recombinant HBeAg at different concentrations.
Detection of recombinant HBeAg with HBeAg/Ab commercial kit.
Production, purification, and assessing the specific reactivity of rabbit anti-HBe-polyclonal antibody
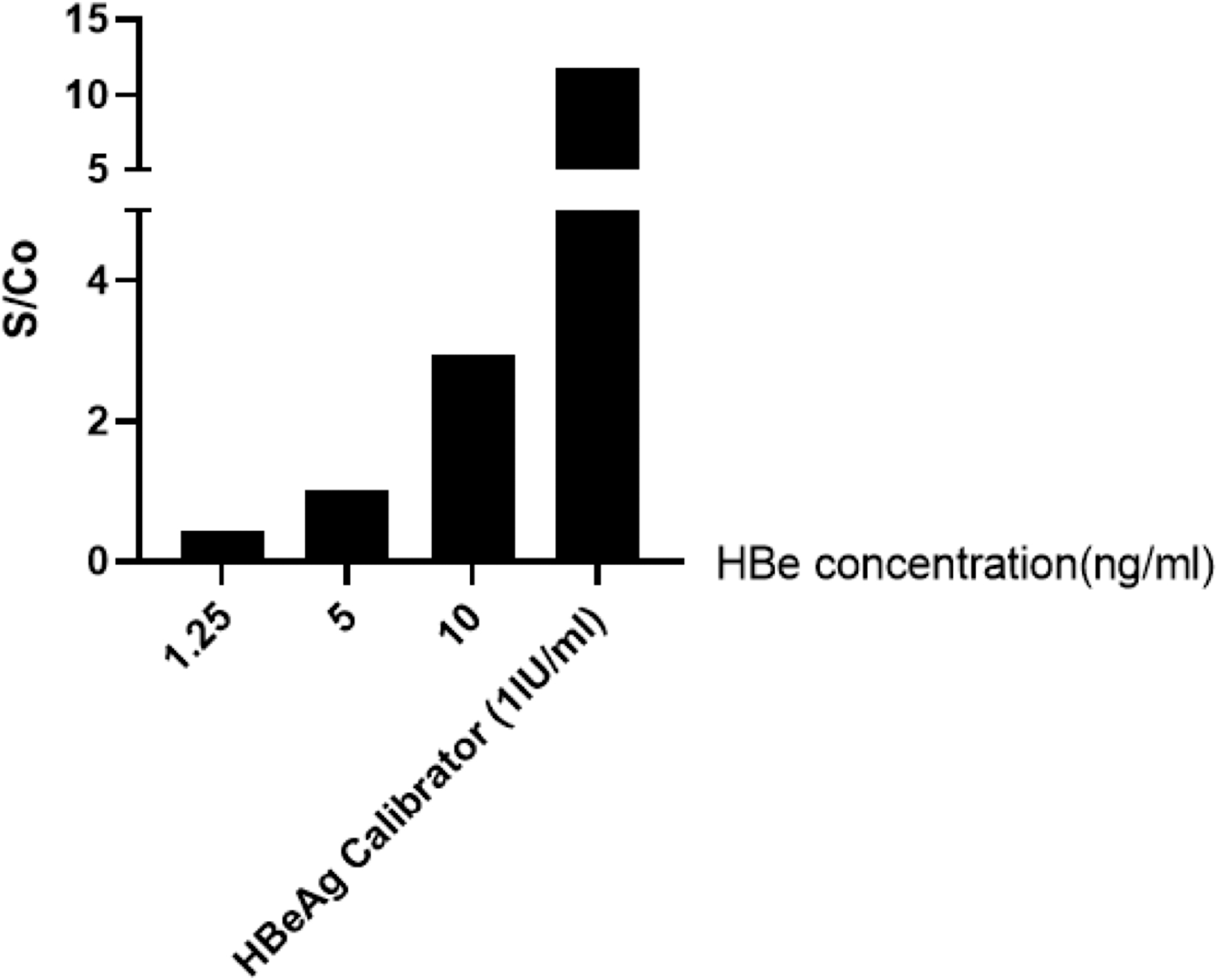
Rabbits were successfully immunized with recombinant purified HBeAg and the specific antibodies were purified by HBeAg-CN-Br-activated Sepharose4B. The specific reactivity of HBeAg-purified anti-HBe polyclonal antibodies with purified recombinant HBeAg was evaluated in ELISA. After biotin conjugation of HBeAg-purified anti-HBe polyclonal antibodies, a sandwich ELISA was designed and our results showed that the optimized assay could detect up to 0.097 ng/mL of HBeAg (Fig. 8A).
Specific reactivity of HBeAg-purified anti-HBe polyclonal antibodies with HBeAg.
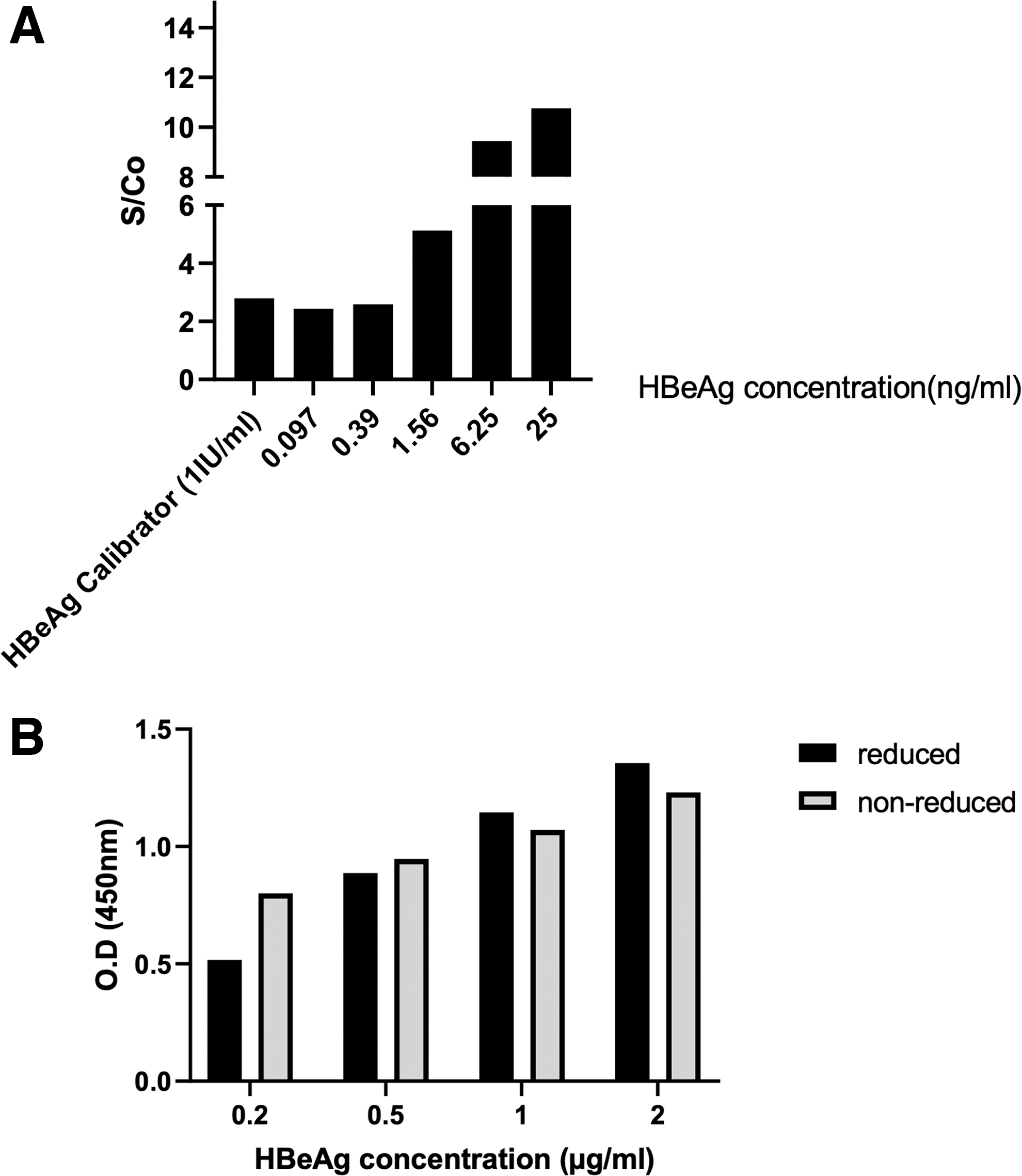
To evaluate the reactivity of anti-HBe polyclonal antibodies to sequential and conformational epitopes, we also performed an ELISA on reduced and nonreduced HBeAg. As Figure 8B shows, anti-HBe polyclonal antibodies showed similar reactivity with HBeAg in reduced and nonreduced conditions at different concentrations.
We also assessed whether anti-HBe polyclonal antibodies generated in rabbit immunized against prokaryotic recombinant HBe has the ability to recognize native HBeAg. Accordingly, HBeAg secreted in the supernatant of HBV-infected HepG2-NTCT cells and HBeAg in the serum of HBV-infected patients used to reach this aim. As Figure 9 shows, HBeAg-purified anti-HBe polyclonal antibodies generated against prokaryotic recombinant HBeAg have the ability to specifically detect native HBeAg in the supernatant of infected HepGE2-NTCP cells and also in the serum of HBV-infected patients.
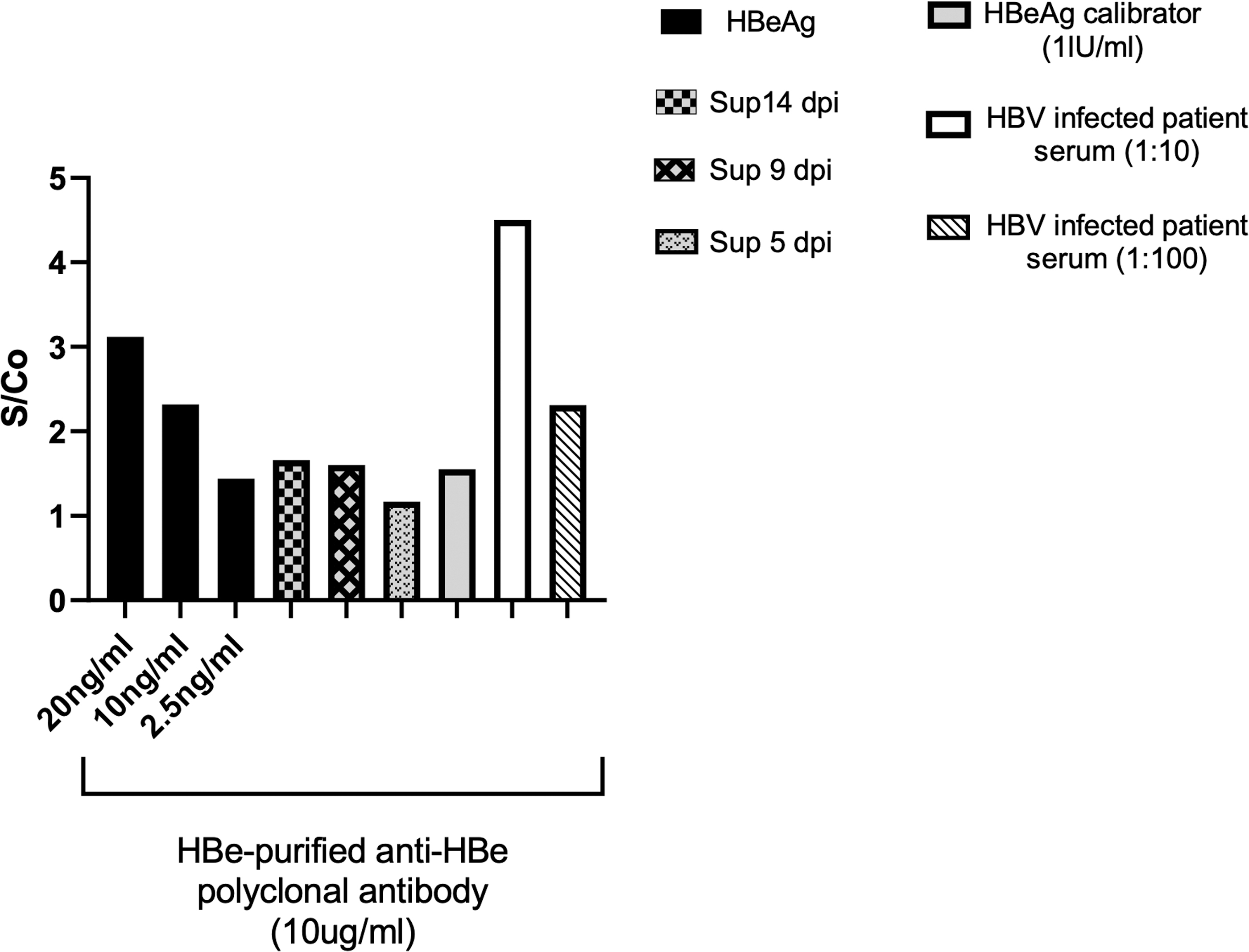
HBeAg-purified anti-HBe polyclonal antibodies could recognize both recombinant and native HBeAg. Ten micrograms per milliliter of HBeAg-purified anti-HBeAg polyclonal antibodies was coated on an ELISA plate. Different concentrations of recombinant HBeAg, as well as supernatant of infected HepG2-NTCP cells, and serum of HBV-infected patients were used. 1 IU/mL of antigen calibrator of HBeAg commercial kit was added as positive control. Biotin-conjugated HBeAg-purified anti-HBe polyclonal antibody was used at 1/2,000 dilution. HBeAg: recombinant HBeAg, Sup 5, 9, and 14 dpi: supernatant of HBV-infected HepG2-NTCP cells at 5, 9, and 14 days postinfection.
Assessing the reactivity of HBcAg-adsorbed anti-HBe polyclonal antibody with HBeAg
To assess whether HBcAg-adsorbed anti-HBe polyclonal antibodies still show cross-reactivity with HBcAg, a sandwich and indirect ELISA was optimized. Our results showed that even after adsorbing cross-reactive antibodies, the majority of HBcAg-adsorbed anti-HBe polyclonal antibodies show cross-reactivity with HBcAg, as checked in both sandwich and indirect ELISA (Fig. 10A, B).
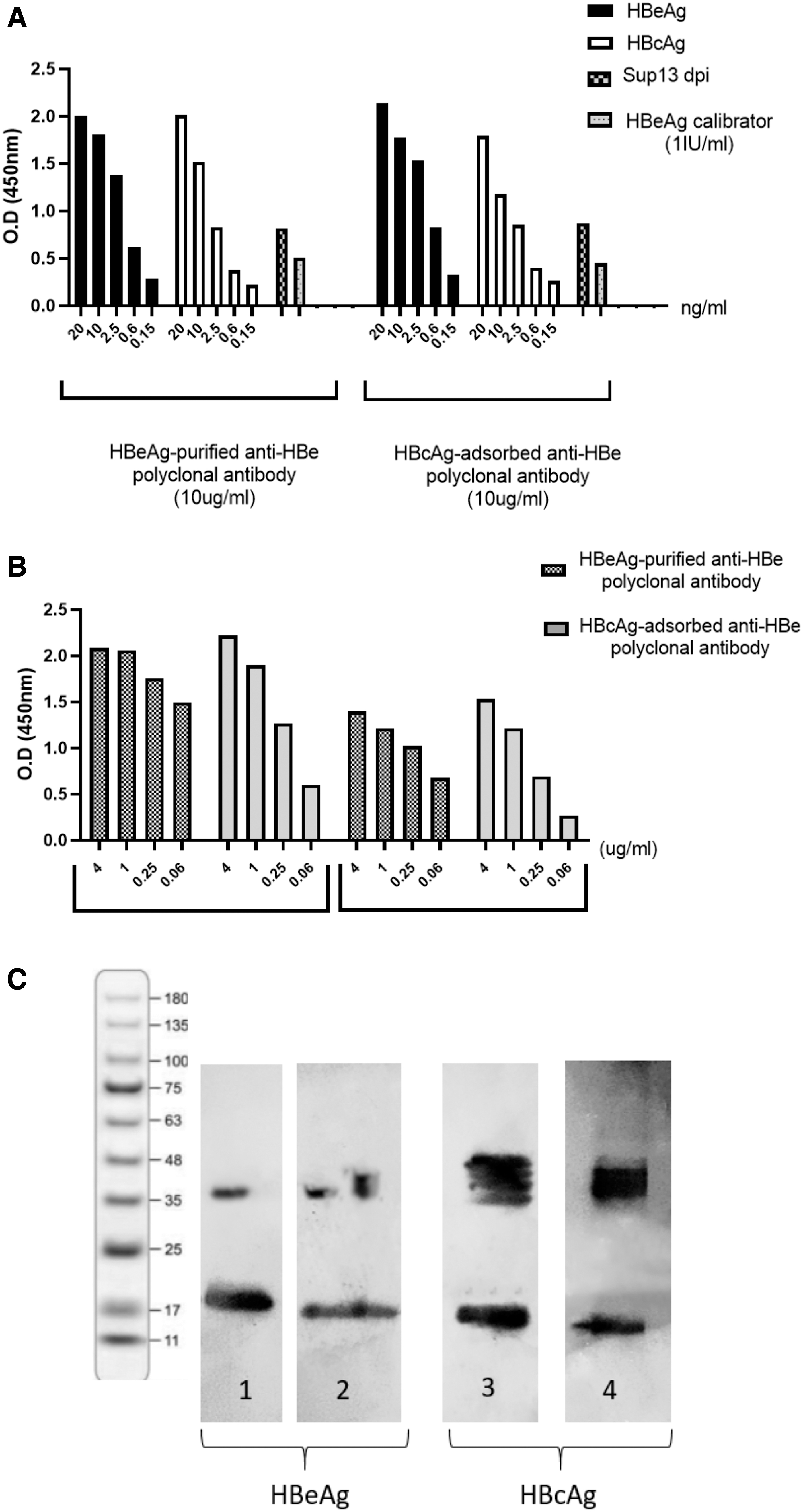
Evaluation of cross-reactivity of HBcAg-adsorbed anti-HBe and HBeAg-purified anti-HBe polyclonal antibodies with HBeAg and HBcAg.
We also evaluated the specific reactivity of HBcAg-adsorbed anti-HBe polyclonal antibodies with recombinant HBe- and HBc- Ag in Western blotting. As Figure 10C shows, HBcAg-adsorbed anti-HBe polyclonal antibodies still showed cross-reactivity with HBcAg.
Discussion
Although HBcAg is the main structural protein of HBV capsid, HBeAg, its function should not be considered “unknown” anymore, shows the immunotolerance/immunomodulation effect in chronic HBV-infected patients (Milich, 2019).
HBc and HBe antigens are derived from the HBV precore/core gene at different in-frame start codons. The first residue of HBcAg is designated as (+1), while HBeAg initiates at residue −29 at the upstream start site of HBcAg, and this precursor further processes at both amino- and carboxyl-terminals, including removal of a 19-residue of leader peptide. HBe and HBc antigens share 149 residues, but HBeAg has an additional 10 residues at its amino termini (aa −10 to −1, SKLCLGWLWG), whose presence correlates with immunological and physical difference between the two antigens (DiMattia et al., 2013).
HBeAg, as an HBV infection biomarker, is an important tool for monitoring chronic HBV infection both in the natural course of infection and during and after treatment. Despite the diagnostic importance of HBeAg in monitoring the status of the infection, current HBeAg detection immunoassays are not optimal due to the cross-reaction with HBcAg. Most of the anti-HBe antibodies used in current HBeAg immunoassays show cross-reactivity with HBcAg (Wang et al., 2021). Since HBV-infected hepatocytes secrete both enveloped virions and also naked HBcAg capsids, the current HBeAg immunoassays could detect both HBeAg and HBcAg in the serum.
Despite high similarity between HBe and HBc antigens, previous studies have reported the generation of monoclonal antibodies, which specifically recognize each antigen with no cross-reactivity with the other one (Sogut et al., 2011; Wang et al., 2021; Zhuang et al., 2017). However, the specific reactivity of HBcAg-adsorbed anti-HBe polyclonal antibodies with HBeAg has not been assessed so far.
In this study, we generated and purified recombinant HBeAg in E. coli and used it to produce anti-HBe polyclonal antibodies. The recombinant prokaryotic HBeAg was further characterized using sera of HBV-infected patients containing anti-HBe antibody. We used HBeAg-purified anti-HBe polyclonal antibodies to design an ELISA for HBeAg quantification, both recombinant and also native HBeAg in the patient serum samples. Next, recombinant HBcAg was generated and coupled to CNBr-activated Sepharose resin. For the first time, we evaluated whether HBcAg-adsorbed anti-HBe polyclonal antibodies could specifically recognize HBeAg or still show cross-reactivity with HBcAg.
In this study, HBeAg (amino acids −10 to 149) and HBcAg (amino acids +1 to 149) gene sequences were amplified from a plasmid carrying complete genome of HBV (pT-HBV1.3) and then inserted downstream of the hexa-histidine tag sequence between NdeI and HindIII sites of pColdI vector. It is noteworthy that we first cloned HBeAg gene in pET22b vector, but the expression level of this protein in different hosts of E. coli was extremely low, and thus, we had to examine the expression level of this protein in pCold1 vector for the first time. Although cloning of HBc particles (amino acids 1–183) in pET22b and expression of this antigen in BL21 (DE3) strain had been previously reported (Bin Mohamed Suffian et al., 2017), the expression level of HBe and HBc antigens cloned in pET22b in our study was extremely low.
Expression level of HBeAg and HBcAg (HBeAg- and HBcAg-pColdI) in six different expression hosts, including BL21 (DE3), Rosetta-gami (DE3) plysS, Origami (DE3), Novablue (DE3), Tuner (DE3), and Shuffle (DE3), was evaluated and the highest expression level of both proteins was detected in BL21 and Rosetta-gami strains (Fig. 3). The overexpressed HBeAg seems similar to a double band, which might be due to the presence of high amounts of HBeAg as a thick band. Another explanation could be due to the degradation of some of the HBeAg to smaller fragments (Fig. 3b, lane 3). Since proper folding of recombinant HBeAg and HBcAg requires formation of intramolecular disulfide bonds between cys (−7) and (61) or intermolecular disulfide bond between cyc (61), respectively, the antigens were expressed in Rosetta-gami, which its oxidative cytoplasm supports the formation of disulfide bonds (Bessette et al., 1999; Nazari et al., 2017; Watts et al., 2011).
The majority of HBeAg was accumulated in inclusion bodies, and thus, lysis buffer containing 6 M urea was used to solubilize it. Following binding of recombinant HBeAg or HBcAg to the Ni-NTA column, the solubilized proteins were refolded by gradual removal of urea from 6 to 0 M during column washing. Unfortunately, this process resulted in loss of the majority of the antigens during elution. Removing 6 M urea during dialysis also led to antigen aggregation. Therefore, 2 M urea was kept in the elution buffer and dialysis buffer to avoid protein aggregation. Other alternatives such as adding glycerol or SDS could not prevent protein aggregation.
Therefore, to avoid HBeAg or HBcAg aggregation during the purification process, the antigens were eluted in a buffer contained 2 M urea and further dialyzed in a buffer containing the same concentration of urea. It is noteworthy that 2 M urea did not interfere with antigen coupling to CN-Br agarose. The total recovery of HBeAg was determined to be 4 mg/L of culture of E. coli Rosetta-gami bacteria.
We further evaluated whether recombinant HBeAg shows structural and immunological similarity with the native one. Accordingly, anti-HBe antibodies in the serum of chronically infected patients recognized recombinant HBeAg in both ELISA and Western blotting (Figs. 5 and 6), implying that recombinant HBeAg maintained its antigenic properties in the prokaryotic system and there is a high similarity between the recombinant and native antigens. Our results were in accordance with previous studies reporting that rHBeAg produced in bacteria had a similar structure to its native form (Schödel et al., 1993; Zhuang et al., 2017). Next, recombinant HBeAg was used to produce rabbit anti-HBe polyclonal antibodies. The animals were successfully immunized with the recombinant HBeAg dissolved in buffer containing 2 M urea, suggesting that urea may not interfere with the animal immune system.
Using HBeAg-purified anti-HBe antibodies, the ELISA system was optimized and was able to detect recombinant HBeAg up to about 0.09 ng/mL. HBeAg-purified anti-HBe antibodies showed similar reactivity with HBeAg in reduced and nonreduced conditions, implying their reactivity with both sequential and conformational epitopes in the recombinant HBeAg (Fig. 8B). Unfortunately, we did not have access to CD-spectroscopy to assess the secondary structure elements, which is one of the limitations of this study.
In addition, HBeAg-purified anti-HBe polyclonal antibodies were also able to detect native HBeAg in serum of HBV-infected patients and also in the supernatant of HBV-infected HepG2-NTCP cells, emphasizing the suitability of prokaryotic system to generate recombinant HBeAg with a similar structure to native one (Fig. 9).
HBcAg-adsorbed anti-HBe polyclonal antibody still showed high cross-reactivity with HBcAg (Fig. 10). Therefore, to develop a sensitive ELISA that does not show cross-reactivity with HBcAg, only anti-HBe monoclonal antibodies with the ability to differentiate the two antigens are crucial.
Footnotes
Acknowledgment
We appreciate Professor Michael Nassal for scientific consultation.
Authors' Contributions
Ms. M.H., Dr. M.M.A., Ms. M.M., Dr. M.H.M., Dr. M.G., Dr. S.G., Ms. K.P., Dr. F.G.-S., and Dr. F.S. are from the Tehran University of Medical Sciences (Tehran, Iran); Dr. S.G. is also from the Kashan University of Medical Sciences (Kashan, Iran); and Dr. F.S. is also from the Avicenna Research Institute, ACER (Tehran, Iran), all where education and research are the primary functions.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the Tehran University of Medical Sciences (grant numbers 98-01-27-42241, 97-02-27-37246, and 97-02-27-37247) and the Iran National Science Foundation (INSF) (grant numbers: 95836480 and 97015900).