
Abstract
Zika virus infections lead to neurological complications such as congenital Zika syndrome and Guillain–Barré syndrome. Rising Zika infections in newborns and adults have triggered the need for vaccine development. In the current study, the precursor membrane (prM) protein of the Zika virus is explored for its functional importance and design of epitopes enriched conserved peptides with the usage of different immunoinformatics approach. Phylogenetic and mutational analyses inferred that the prM protein is highly conserved. Three conserved peptides containing multiple T and B cell epitopes were designed by employing different epitope prediction algorithms. IEDB population coverage analysis of selected peptides in six different continents has shown the population coverage of 60–99.8% (class I HLA) and 80–100% (class II HLA). Molecular docking of selected peptides/epitopes was carried out with each of class I and II HLA alleles using HADDOCK. A majority of peptide–HLA complex (pHLA) have HADDOCK scores found to be comparable and more than native–HLA complex representing the good binding interaction of peptides to HLA. Molecular dynamics simulation with best docked pHLA complexes revealed that pHLA complexes are stable with RMSD <5.5Å. Current work highlights the importance of prM as a strong antigenic protein and selected peptides have the potential to elicit humoral and cell-mediated immune responses.
Introduction
Zika virus is one of the most perilous flaviviruses associated with the flaviviridae family of arboviruses. Neurological disorders, including fetal microcephaly, Guillain–Barré (gee-YAH-buh-RAY) syndrome, and ocular abnormalities in humans, are caused by the Zika virus. Since January 2007, evidence of autochthonous mosquito-borne transmission of Zika virus infection has been reported in 39 countries and territories (WHO, 2019). The Zika virus is of so much concern lately, not from the infection itself but its consequences, particularly in pregnant women. Susceptibility to Dengue, Zika, and other arboviruses is progressive in different geographical regions of India due to their dependence on mosquitoes, especially Aedes (Ae.) aegypti in particular (Khaiboullina et al., 2018). Apart from that, Zika virus can also be transmitted by Ae. africanus, Ae. furcifer, Ae. apicoargenteus, Ae. Luteocephalus, and Ae. vitattus, mosquito species (Kazmi et al., 2020).
Infection with all types of Zika strains, including African (MR766), Asian (FSS13025, PF/2013/KD507), and American (ZIKA-BR/215, KU527068) (Dang et al., 2016), can result in microcephaly, a disastrous condition that causes an underdeveloped brain, small head size, developmental delay, developmental failure, learning disability, short stature, hearing loss, and intellectual disability in the newborn.
Zika primarily invades the astroglial and microglial cells in adults and neural progenitor cells (NPCs) in the embryonic brain, replicate within, and endure neuropathic damage and congenital malformations (Horibata et al., 2021; Tang et al., 2016). Zika virus has [ssRNA(+)] genome, roughly in size 10.7 kilobases, which are flanked through two terminals of noncoding regions (NCR), that is, 5′NCR (107 nt) and 3’NCR (428 nt) (Faye et al., 2014). The genomic RNA translates into a single polyprotein within the host cell (Li et al., 2017). The polyprotein further cleaved by cell and host protease into viral structural protein anchor Capsid (C), Envelop (E), Precursor membrane (prM), and Nonstructural (NS) proteins NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. The structural protein, prM has shown to be critical in virus contagion due to its prominence in mature virion assembly, and inhibition of the protein debilitates the pathogenicity of the virus (Yoshii et al., 2012). The prM protein has been found as a prominent target for cell-mediated immune response, especially for CD8+ T cells of human (Grifoni et al., 2017) and mice (Pardy et al., 2017).
A study based on crossreactive immune response shows that the protective CD8+ T cells recognize the epitopes of, mainly prM, E, and C proteins, in naive Zika infections (Ngono and Shresta, 2019). It has been found that ZIKV-specific antibodies elicit an immune response against the prM and E proteins (Screaton et al., 2018). In Zika virus, N-linked glycosylation (N-glycan) is found in both prM and E proteins (Sirohi et al., 2016). It has been found that the lack of prM N-glycosylation site favors the incorrect folding of prM protein that impairs the expression and secretion of E protein leading to abolishment or low amount of infectious virus particle release from the host cell endoplasmic reticulum to plasma membrane (Gwon et al., 2020), thus the significance of prM protein increases in Zika virus cycle. The prM protein is the part of subviral particles and virions, thus it is a primary target of vaccine development (Hasan et al., 2018). The prM protein together with E protein pertains to ChimeriVax technology to develop vaccines for different flaviviruses (Guy et al., 2010).
A vector-based vaccine (containing prM proteins along with E and NS1 proteins) was found to be able to induce T cell immunity and Zika-specific antibodies in BALB/c mice (Li et al., 2018). It has also been reported that antibodies against the prM protein can be used as a serologically specific marker to identify Zika infection in areas where Zika is coinfected with other flaviviruses (Hsieh et al., 2021).
Vaccines based on different platforms, plasmid DNA (Tebas et al., 2021), mRNA (Gaudinski et al., 2018), and purified inactivated virus (Modjarrad et al., 2018) vaccines were in clinical trials but no reliable vaccine has been formulated to date. In this contrast, immunodominant peptide-based vaccines represent a more reliable, facile, and protected direction for the persistent immune response against pathogens. Peptide vaccine constructs against various cancers, such as breast cancer (Dillon et al., 2017), lung cancer (Suzuki et al., 2013), prostate cancer (Obara et al., 2017), and myeloid leukemia (Qazilbash et al., 2017), showed encouraging results in clinical trials. A multipeptide vaccine GX-301 for prostate and renal cancer has exhibited good T cell immune response with no adverse effects in the clinical phase I trial (Fenoglio et al., 2013).
Vacc-flu, peptide-based vaccine has been tested for its efficacy in the mouse model. The study concludes that the vaccine can elicit influenza-specific antibodies and cell-mediated immune response (Herrera-Rodriguez et al., 2018). Recently multipeptide-based vaccine, P-pVAC-SARS-CoV-2 (NCT04546841), is in phase I clinical trial for combating COVID-19 (Hilpert and Apostolopoulos, 2021).
Several research studies have found that peptides obtained using high-throughput immunoinformatic approach have proven to produce promising results at the in silico level validation (Fatima et al., 2022; Heidary et al., 2022; Kaushal et al., 2022). Computationally, highly conserved fragments of viral proteins, their characteristics, and their in vitro validation (Díaz-Gómez et al., 2020; Jain and Baranwal, 2021; Sharma et al., 2021) were identified, indicating widespread use of this approach and elevating the peptide vaccine development process (Soleymani et al., 2021). The peptides predicted by these approaches showed potential in ELISA assay, which proves their usefulness for immunological studies (Li et al., 2022).
In this proof of concept, recently in vivo analysis of the first computational design peptide-based vaccine of SARS-CoV-2 was done successfully (Martínez et al., 2022). There have been reports, which demonstrated that the association of prM protein with capsid (C) or Envelop (E) protein facilitates therapeutic development, including DNA, recombinant virus vectors, mRNA vaccines, and VLPs (Lin et al., 2018). A better antigenicity of any protein depends on its behavior and many parameters.
Phylogenetic and mutational analysis reveals that protein has strong conservancy with minimum variance. Furthermore, three strong immunogenic and conserved peptides were obtained, which were enriched with T and B cell epitopes. Current prediction included different epitope prediction algorithms and 2,915 human alleles of class I MHC, and 687 human alleles of class II MHC. The selected peptides have good restriction of HLAs presented to all over the world showing 60–99.8% (class I HLA) and 80–100% (class II HLA) population coverage. Molecular docking analysis with selected peptides/epitopes and 10 each of class I and II HLA showed good HADDOCK scores of peptide–HLA (pHLA) complex. On applying MD simulations up to 100 ns, they showed RMSD values <5.5 Å indicating the good binding strength and stability of the selected peptides.
Materials and Methods
Construction of dataset
Due to less number of Zika prM protein sequences in repository sources, the polyprotein sequences of the virus were taken using the search details; Zika virus, as an organism; polyprotein, as protein name; sequence length as 3,423 amino acids; and sequence releasing date up to July 31, 2022 from advance search builder option of NCBI. The unique and human host sequences were separated and the sequence of prM protein was identified and fetched from each polyprotein sequence.
Phylogenetic analysis
The phylogenetic relation among the sequences was carried out using the Maximum Likelihood method and JTT matrix-based model (Jones et al., 1992) through the MEGAX suite (Kumar et al., 2018). Clustal Omega (1.2.4) (Sievers et al., 2011) was used for multiple sequence alignment (MSA). The input was given as an MSA result of sequences in FASTA format.
Mutation and conservation analysis
The MSA result was analyzed using the BioEdit tool (Hall, 1999) for locating mutations. The mutational frequency at a particular position was calculated by taking the ratio of the number of sequences found to be mutated at that position and the total number of sequences. AVANA (Antigen Variability ANAlyzer) tool (Miotto et al., 2008) was used for finding conserved peptides above 90% from the prM protein sequences. Minimum and maximum peptide lengths were set as 9 and 25 residues, respectively. The protein blast search was done using NCBI pBLAST (Altschul et al., 1997) against a nonredundant protein sequence of homo sapiens (taxid: 9606) to select the conserved fragments, which are not resembling to human proteins. The assessment was based on matching a minimum of seven identical continuous amino acids to human proteins.
Identification of functional domains and superfamilies
The functional domains of the proteins were identified using CATH server (Dawson et al., 2017), taking into account the domain contribution to the functions of the protein. InterProScan tool (Blum et al., 2021) server was used for verification. FASTA sequence of prM protein was given to the server for investigation of protein hierarchy.
CD8+ and CD4+ T cell epitope prediction
Epitopes for CD8+ T cells (Class I HLA-binding epitopes) were predicted with NetMHCpan 4.0 (Jurtz et al., 2017), NetCTLpan 1.1 (Stranzl et al., 2010), and NetMHCstabpan (Rasmussen et al., 2016). The threshold values in NetMHCpan 4.0 server were set as percentage rank 0.5 for strong and 2.0 for weak binders by selecting binding affinity (BA) and Eluted Ligand (EL) parameters. NetCTLpan 1.1 server showed epitope identity at −99.9 and 1.0 threshold, weighting on C-terminal cleavage and the transporter associated with antigen-processing (TAP) efficiency were specified as 0.225 and 0.025, respectively. The NetMHCstabpan server thresholds were considered as 0.5 and 2.0 percentage ranks for strong and weak binding stability of peptides, respectively. CD4+ epitopes (Class II HLA-binding epitopes) were predicted by the NetMHCIIpan 3.2 server (Jensen et al., 2018) with 2.0 and 10.0 thresholds for strong and weak binders correspondingly, without excluding offset correction.
The Stabilization Matrix Alignment Method (SMM)-alignment (Nielsen et al., 2007) server of the IEDB analysis resource and TEPITOPEpan (Zhang et al., 2012) were used as the other two prediction tools for CD4+ epitopes by selecting human alleles of HLA-DP, DQ, and DR locus.
B cell epitope prediction
Continuous B cell epitopes were predicted through ABCpred server (Saha and Raghava, 2006b). The server used a threshold value of 0.51 with default window size for accurate and sensitive analysis. The results were obtained based on the amino acid propensity scale and HMM (Hidden Markov Model) algorithm.
Covered HLA molecules and conservancy of peptides
The HLA restricted by the epitopes presented within the peptides were estimated by counting the unique bound HLA alleles of all supertypes. The conservation of the final peptides in all 345 sequences was analyzed through the IEDB resource (Bui et al., 2006). The sequences of the peptides were submitted to the server with the Epitope Linear Sequence Conservation option at > = 90% sequence identity threshold.
Assessment of Antigenicity, toxicity, and allergenicity property
VexiJen 2.0 server (Ong et al., 2020) was employed to assess the antigenic potential of designed peptides. Peptide sequences were submitted separately to the server. The target organism was selected as a virus with a 0.4 threshold value. Possible health risk factors, toxicity, and allergenicity of all the peptides were evaluated through ToxinPred (Gupta et al., 2013), AlgPred (Saha and Raghava, 2006a), and AllergenFP 1.0 (Dimitrov et al., 2013) servers by providing input as peptide sequences with the default threshold values.
Estimation of physiochemical properties
Various physical and chemical properties of peptides were analyzed through the ProtParam tool (Gasteiger et al., 2005). The calculated parameters included molecular weight, isoelectric point, instability index, aliphatic index, and grand average of hydropathy (GRAVY).
Population coverage analysis
Estimating population coverage was achieved using the IEDB analysis resource population coverage tool (Bui et al., 2006). Analysis was done separately for CD8+ and CD4+ T cell epitopes for 115 countries and 16 different geographical areas with 21 diverse ethnicities. The query measure was selected, including area, country, and ethnicity with the entire world, including 16 geographical areas. Epitope sequences were given, and their HLA restriction alleles were selected from the given list for analysis.
Molecular docking and simulation
The interactions between peptides containing multiple CD8+ and CD4+ T cell epitopes, and various HLA molecules of class I and II were analyzed through docking study by using the HADDOCK (High Ambiguity Driven protein–protein DOCKing) server (Van Zundert et al., 2016). A total of twenty, peptide-bound HLA structures were retrieved from the PDB database representing different HLA class I and II supertypes. PyMOL tool (Schrödinger and DeLano, 2020) was used for separating HLA and peptides structural (.pdb) files, and resultant peptides and HLAs were used as native peptides (NP) and receptors, respectively. The tertiary structures of identified peptides were formed through Modeller (Webb and Sali, 2016). GalaxyRefine web server (Heo et al., 2013) was utilized for the refinement of structures files. Energy minimization of structures was done through Chimera (Pettersen et al., 2004).
The three best docked pHLA complex of HLA class I and II were subjected to molecular dynamics (MD) simulation to analyze the stability of docked complex (pHLA) through GROMACS version -2021.3. CHARMM–GUI interface using CHARMM27 force field was used for generating topology and coordinates of molecules. Energy minimization of and two-step equilibration were carried out for each complex. The full simulation was done with 100 ns for all the subjected molecules.
Protein structure modeling
The structure of prM protein is not available in the PDB database, so the protein structure was modeled using Modeller v9.24 (Webb and Sali, 2016). The reference sequence of Zika prM protein was queried to identify homolog sequences by BLASTp. The obtained .two hits, Cryo-EM structure of Spondweni virus prME (PDB ID: 6ZQI_B; 99% of query coverage; E-value of 1e-86), and prM protein of Binjari virus (PDB ID: 7L30_a; 96% of query coverage; E-value of 9e-36) were found with complete side chains used as the template for model formation. All the required files, including query sequence file, template file, and other script files, were prepared and initialized in Modeller script files (
Docking of prM protein with immune receptor
Docking between prM protein and TLR3 was performed by HADDOCK server (Van Zundert et al., 2016). PDB structure of TLR3 was retrieved from PDB database, which was refined by GalaxyRefine web server (Heo et al., 2013), and minimization done through Chimera (Pettersen et al., 2004). Active residues of prM protein and TLR3 were given with structure files in the server for docking.
Results
Construction of dataset
Six hundred two sequences, including reference sequence (RefSeq), YP_009430297.1 (Mlakar et al., 2016) of Zika virus polyprotein were retrieved according to the search details provided in protein advance search builder of NCBI. All the obtained sequences were assessed using unique sequence generator tool, and a total of 345 nonredundant and complete polyprotein sequences were obtained. All the sequences were scrutinized, and a stretch of amino acids from 123 to 290 positions (prM protein sequence position) was fetched from each complete polyprotein sequence. Finally, the dataset of 345 sequences of prM protein was constructed for further analysis.
Phylogenetic analysis
The phylogenetic tree shown in Figure 1 was constructed with 1,000 bootstrapping replications of maximum likelihood at the highest log likelihood (−912.27) for revealing the evolution history of prM proteins. The analysis involved 168 site alignment lengths of 345 prM protein sequences. The tree was rooted with YP_009430297.1 RefSeq (Brazilian taxon) as the most recent common ancestor. The branch length and topology of the tree infer the probability of the resemblance and the divergence of sequences. Clusters were formed by compressing the sub-trees of clades with similarity. Clusters were named from cluster-A to cluster-X (data given in Supplementary Table S1). Maximum taxons showed proximity with the reference sequence. The clades within the clusters showed more distantly from reference sequence because of the presence of additional nodes between the root and clusters, indicating change in amino acids (genetic distance) of clades. The phylogeny of the prM protein shows that the protein retained its conservation in the years of peak outbreak (2015 to 2016), except for some variations.

Phylogenic tree of prM protein sequences of Zika virus up to 2022. The tree presents evolution history of prM protein. Clades within the subtrees grouped into clusters showing divergence from reference sequence. prM, precursor membrane.
Identification of mutation and conserved fragments
Mutations in viral proteins control their evolution, which is important for counteracting pathogens. Mutation analysis revealed the number of mutated amino acids present in the prM protein. The multiple sequence alignment result of Clustal Omega was used for finding the mutations in prM protein sequences. Total, 49 unique mutations were identified at 46 positions in 344 prM protein sequences using RefSeq, YP_009430297.1 through BioEdit. More than one amino acid change was found at the three different locations (N17→S, K), (R89→W, Q), and (Y167→H, C). The substitution of Asparagine with Serine (N17→S) occurred in 72 sequences out of 344 with a frequency of 0.209, but substitution of Asparagine with Lysine (N17→K) was found in single sequence (Fig. 2). In addition, Threonine substitution with Alanine (T74→A) in 12 sequences with frequency 0.03 and Valine substitution for Alanine (A01→V) in 8 sequences with frequency 0.02 was obtained. The frequency of mutations at other positions was equal to or less than 0.01. The investigation clarified that the protein has an overall low mutation rate, as shown in Figure 2.
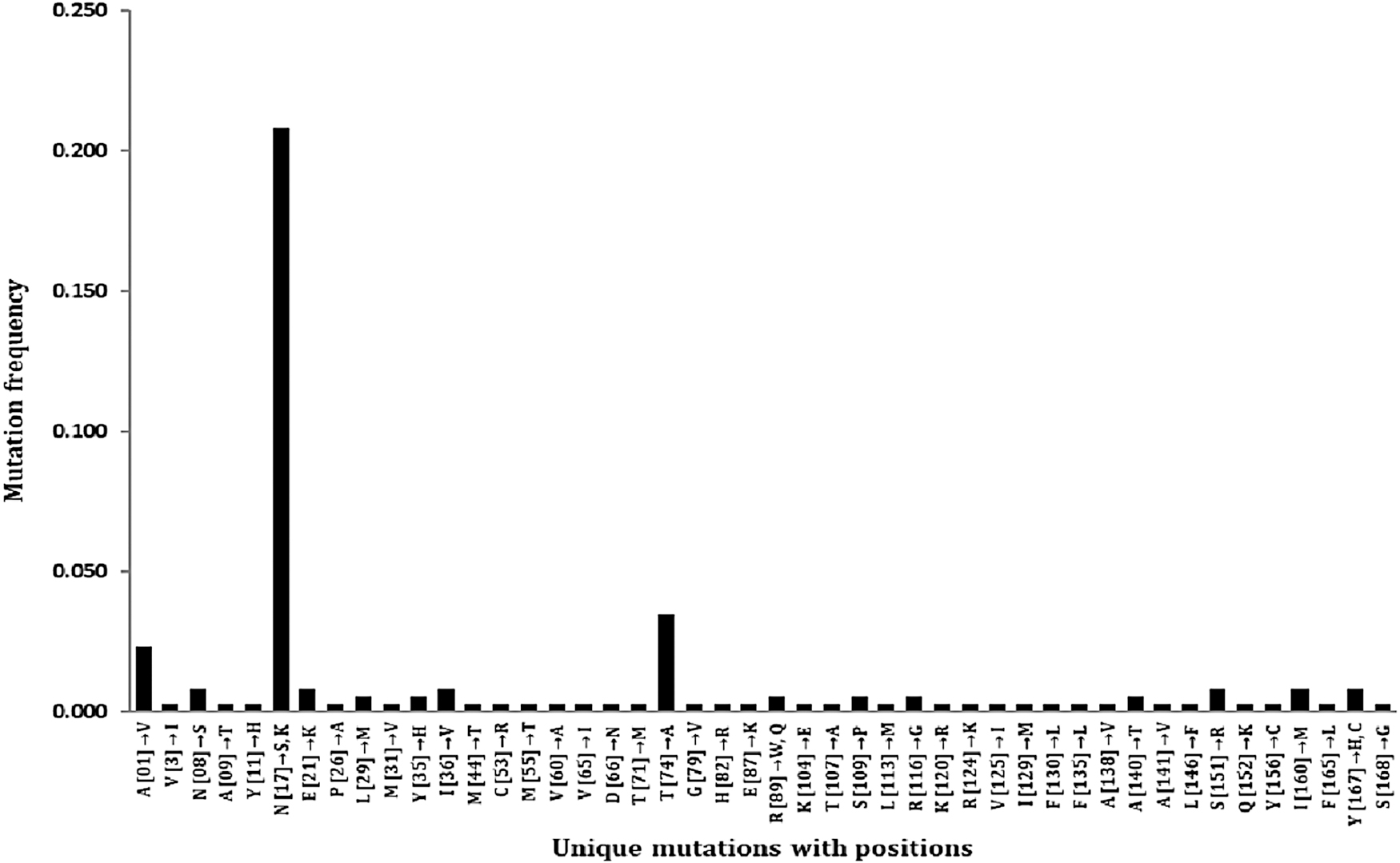
The graph has shown the mutational frequency of amino acids at 46 positions in the prM protein of the Zika virus. Among the 344 sequences, the substitution, S17N, was found in a maximum number of sequences with 0.20 frequency, and T74A has exhibited the second most variation with 0.03 frequency.
The AVANA tool measured the positional conservations in aligned protein sequences through information entropy. Two conserved fragments (1–16 and 18–168) in prM protein were found at 98.46% conservation. Only a single position, 17 having N, S, and K variants in multiple sequences, showed position entropy higher than others.
The pBLAST with human protein resulted in four small fragments, SAYYMYL, ARRSRRA, AWLLGSS, and LVMILLI of seven linear amino acids, which are identical to the following human proteins, including (i) TCR gamma alternate reading frame protein isoform 2 (Sequence ID: NP_001003806.1), (ii) Sortilin-related VPS10 domain containing receptor 3, isoform CRA_a (Sequence ID: EAW49589.1), (iii) CKLF-like MARVEL transmembrane domain containing 4, isoform CRA_a (Sequence ID: EAW83036.1), and (iv) Olfactory receptor 2T2 (Sequence ID: NP_001004136.1), respectively. After removing these identical segments, two conserved fragments, positions (18–88) and (94–142), were obtained, which were used to determine further for immunogenic epitopes.
Identification of functional domains and superfamilies
Functional domain identification was done to locate the conserved fragments and mutations. Domain search clarified that the prM protein consists of two functional domains with their different functionalities, Flavivirus polyprotein propeptide (Flavi_propep), positions 6–84 and Flavivirus envelope glycoprotein M (Flavi_m), interval 94–168, shown in Figure 3. The InterProScan tool and the CATH server also identified the exact domains.
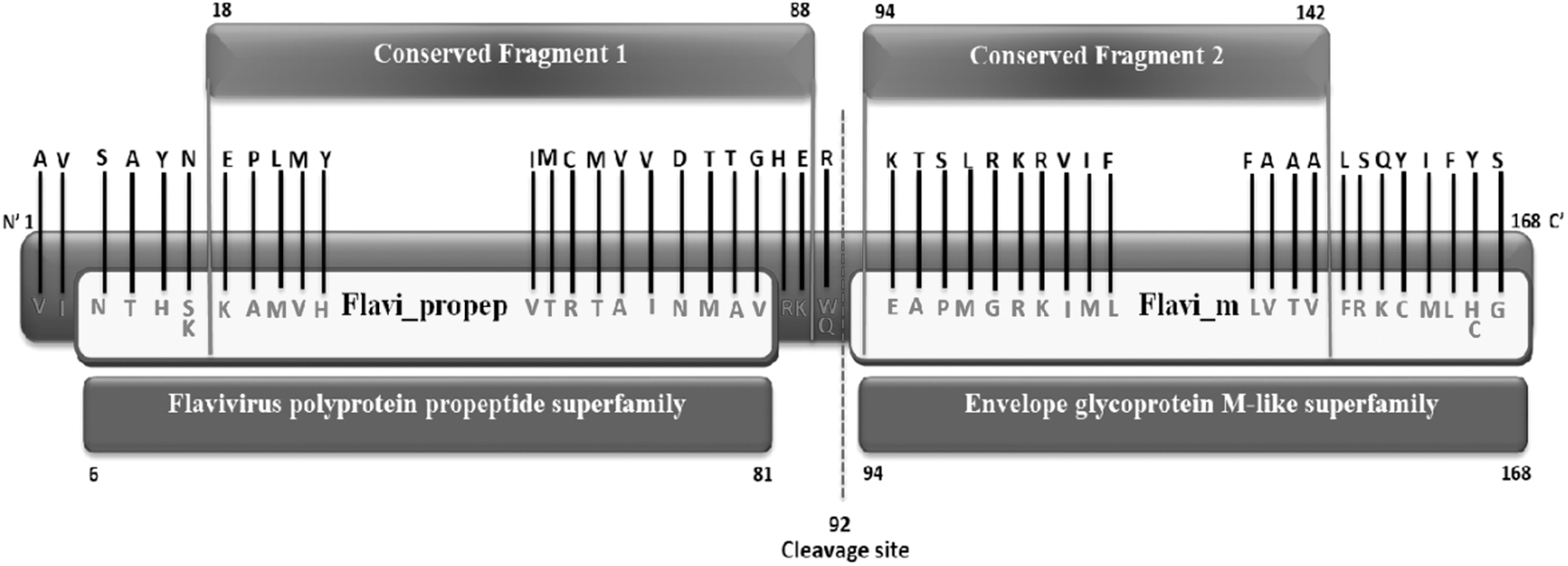
Schematic representation of functional domains of prM protein, Flavi_propep, and Flavi_m. Substituted amino acids are marked within the domains and the conserved fragments are also marked.
The Flavi_propep domain consists of ∼90 amino acid propeptide region. This domain is critical for the protection of premature cleavage of the prM protein (Cox et al., 2015). This domain (CATH ID: 3c5xC00) was found to belong to the Flavi_propep superfamily. In addition, the Flavi_m domain (CATH ID: 6co8B00) belonged to the Flavivirus envelope glycoprotein M-like superfamily. A transmembrane-anchored region was confirmed in the Flavi_m domain, which is required for viral entry into the host cell membrane. Analysis of the superfamily suggested that prM proteins are related to the protein-binding molecular function. It was confirmed through the gene ontology annotation term (GO: 0005515) that the prM protein has binding properties.
Two selected conserved fragments (18–88) and (94–142), were found implicit in the functional domains (Fig. 3). Twenty substituted amino acids were found in the Flavi_propep domain, whereas in the Flavi_m domain, the number of substitutions was found to be 23 through mutational analysis, displayed in Figure 3, but the frequency of these amino acid changes was <0.01.
CD8+ and CD4+ T cell epitope prediction
Covering the HLA-A, HLA-B, and HLA-C supertypes, 2915 human alleles were used in each server for CD8+ T cell epitope prediction. Both conserved fragments (18–88) and (94–142) yielded 33, 25, 30 unique epitopes from NetMHCpan versions 4.0, NetCTLPan 1.1, and NetMHCstabPan 1.0, respectively. Out of all these epitopes, 19 epitopes were found, in the results of all three servers (Table 1). Similarly, 687 human alleles, including HLA-DR, HLA-DQ, and HLA-DP isotypes, were used to predict CD4+ T cell epitopes. Servers, NetMHCIIpan3.2, SMM-align, and TepitopePan predicted 20, 38, and 24 unique epitopes, respectively (Table 1). Out of the epitopes obtained by the three servers, 13 epitopes were found to be common (Table 1). The common CD8+ and CD4+ T cell epitopes were looked for overlapping them. Five and four peptide fragments containing multiple CD8+ and CD4+ T cell epitopes were obtained. From these peptide fragments, three peptides were selected, which contains both CD8+ and CD4+ T cell epitopes (Tables 1 and 2).
Peptides Containing Multiple T Cell Epitopes of Zika Virus prM Protein
Peptides Containing Multiple T and B Cell Epitopes of Zika Virus Precursor Membrane Protein
B cell epitope prediction
Sequential B cell epitopes are essential for generating neutralizing antibodies and may also enhance the cytotoxic lymphocyte (CTL) response through antibody-dependent cell-mediated cytotoxicity. The ABCpred server predicted nine immunodominant continuous B cell epitopes in the selected peptides based on amino acid propensity scales. The results showed B cell epitopes were present in the selected peptides. Two epitopes in peptide MP1, 3 in MP2, and 4 in MP3 were confirmed (Table 2).
Covered HLA molecules and conservancy of peptides
Binding the maximum number of HLA molecules to an antigen indicates broad immunogenic capacity of the antigen that favors T cell activation. In this analysis, epitopes of selected peptides were looked for the number of unique HLA alleles they were predicted to be.
This study found that all the epitopes predicted were binding with large number of HLA molecules. Table 3 shows the total number of HLA alleles used in the prediction and the number of unique alleles restricted by the epitopes of CD8+ and CD4+ T cells in the respective peptides, including both the MHC classes. The MP2 was found to be the most HLA-binding peptide with predominantly 1,495 HLA class I and 263 HLA class II alleles. MP1 and MP3 displayed good binding to a large number of HLA-A (601) and HLA-C (587) alleles, respectively. Similarly, the HLA-DR alleles (236) with MP1 and (215) with MP3 were found to be the most interacting. In the conservation analysis, selected three peptides showed above 99% conservation with all 345 sequences of the prM protein. Peptides, MP1 displayed 99.6%, and MP2 and MP3 led 100% conservation percentage.
Number of Unique HLA Molecules Bound to Peptides Containing Multiple Epitopes
Assessment of antigenicity, toxicity, and allergenicity property
Selected peptides were analyzed to be suitable protective antigens. The VexiJen 2.0 server expressed the antigenicity of the concluded peptides in the form of their antigenic score. Peptides MP1, MP2, and MP3, with antigenic scores of 0.8, 1.09, and 0.46, respectively, were found to have good antigenicity. According to the assessment of toxicity and allergenicity through ToxinPred, AlgPred, and AllergenFP 1.0 servers, peptides were all found to be nontoxic and nonallergenic.
Estimation of physiochemical properties
The computationally deduced physical and chemical features of selected peptides are expressed in Table 4. The values of isoelectric points (pI) of peptides MP1 and MP3 indicated basic nature, whereas peptide MP1 showed acidic character. At these pI values, the solubility of peptides is typically minimal. Peptides MP1 and MP2 had an instability index of less than 40, indicating good stability of the peptides. The Aliphatic index values indicated the relative volume occupied by side chains (Valine, Alanine, Leucine, and Isoleucine), a factor for their thermostability. Selected peptides were found to have aliphatic side chains, showing their aliphatic propensity and thermostable character. The GRAVY scores indicate the hydropathic (relative hydrophobicity or hydrophilicity) nature of peptides. Negative and below zero values of the GRAVY index pointed out that all peptides were hydrophilic. The results showed that selected peptides had good physicochemical properties, enabling them to be considered relevant antigen candidates.
The physicochemical Characteristics of the Final Peptides Are Displayed Below
Population coverage analysis
HLA molecules are the most polymorphic molecules in the human genome. The binding of different epitopes with HLA alleles expresses the substantial effectiveness of antigen on the population. Population coverage results detected the immune significance of selected peptides through particular epitopes in the various worldwide areas. The population coverage of the CD8+ and CD4+ T cell epitopes was measured by taking their average population coverage by combing geographical areas of each continent. The calculation of scores includes populations of related geographical areas, countries, and ethnicities in their respective continents. All CD8+ epitopes showed 60% to 99.8% population coverage through the HLA alleles present in different populations (Fig. 4A). Besides this, the CD4+ epitopes expressed 80% to 100% coverage scores for populations of all the continents (Fig. 4B). Result of the evaluation clarified that both the T cell epitopes cover maximum HLA genotypes in Asia, North America, Australia, Europe, South American, and African populaces.
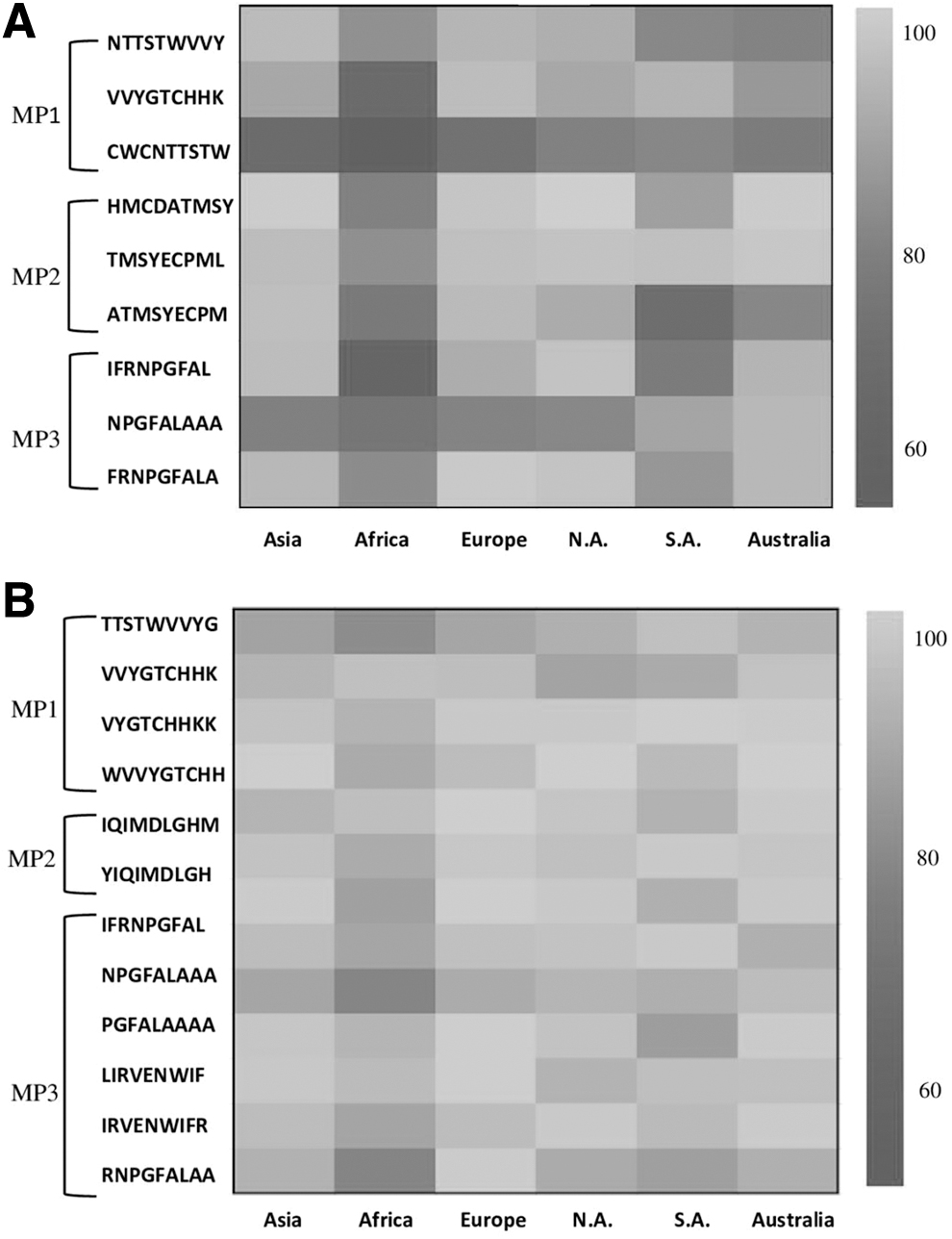
Population coverage by the
Molecular docking of peptides
Efficient interactions between peptides and HLAs are critical for mediating T cell response. A previously done population coverage analysis of the selected peptides has revealed the peptides capable of restricting a large number of HLAs in the entire world, which can be expected to generate a good immune response. Collectively 20 (10 for each HLA class) different peptide–HLA (pHLA) complexes covering diverse HLA supertypes were retrieved from PDB database and peptides and HLA structures were separated.
The separated peptides were used as NP or test control; they docked first and then the selected peptides. Due to the narrow size of the binding groove in the heavy chain, the class I HLA molecules bind with shorter peptides (8–9 mer length), and the class II HLA molecules bind with extended peptides (15–20 mer length) due to the spanning edges of binding groove. Hence CD8+ T cell epitopes (E1 − E9) docked with class I HLA and selected three peptides (MP1 − MP3) as such with class II HLA. The resultant HADDOCK scores of CD8+ and CD4+ T cell peptides are shown through heatmaps in Figure 5A, B, respectively (the value of RMSD and HADDOCK scores given in Supplementary Tables S2 and S3).
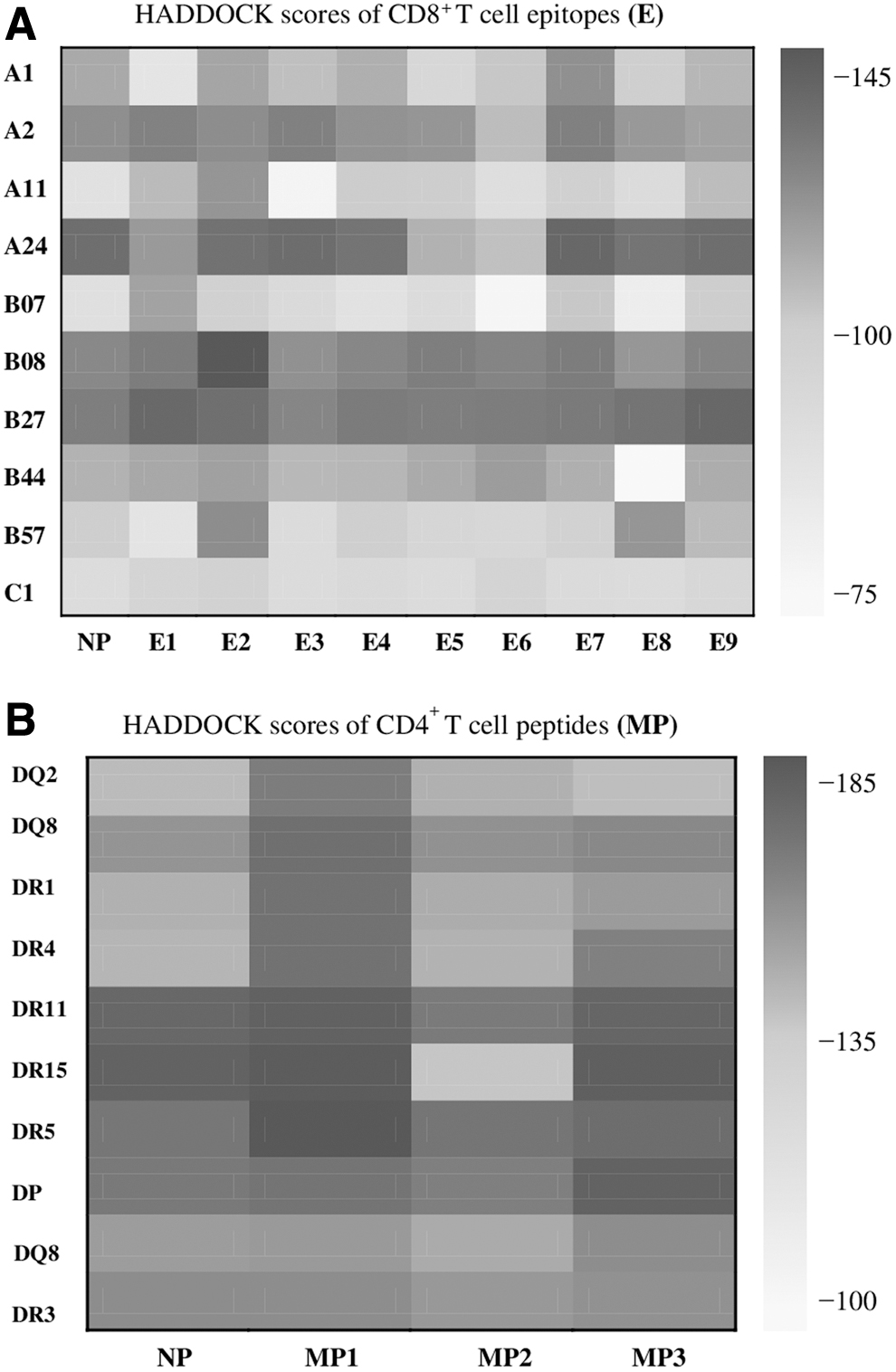
Heatmaps of HADDOCK scores of docked
More negative HADDOCK scores represented better binding, and the RMSD values are calculated according to contacted interfaced residues in case of protein–peptide docking (Trellet et al., 2015). Mostly CD8+ T cell epitopes found binders to HLA molecules. Of these results, three peptides E2, E7, and E9 showed the best HADDOCK scores −148.8, −137.8, and −137.6 for docking with HLA B08, A24, and B27, respectively, and their RMSD values observed were 0.1Å, 0.2Å, and 0.2Å. Similarly, CD4+ T cell peptides MP1 and MP2 showed strong binding with class II HLA-DR5, achieving HADDOCK scores of −187.2 and −170.5, and peptide MP3 binding with HLA-DR15 scored −181.2. The RMSD values, 0.1 Å, 0.2 Å, and 0.3 Å, were obtained for these peptides. The HADDOCK scores of all these selected docked peptides were nearly above native peptide scores and their RMSD values were near native. The binding poses of three best docked peptides of CD8+ and CD4+ T cell are displayed in Figures 6 and 7 by (A), (B), and (C), respectively (data of hydrogen bonds and active residues are given in Supplementary Table S4).

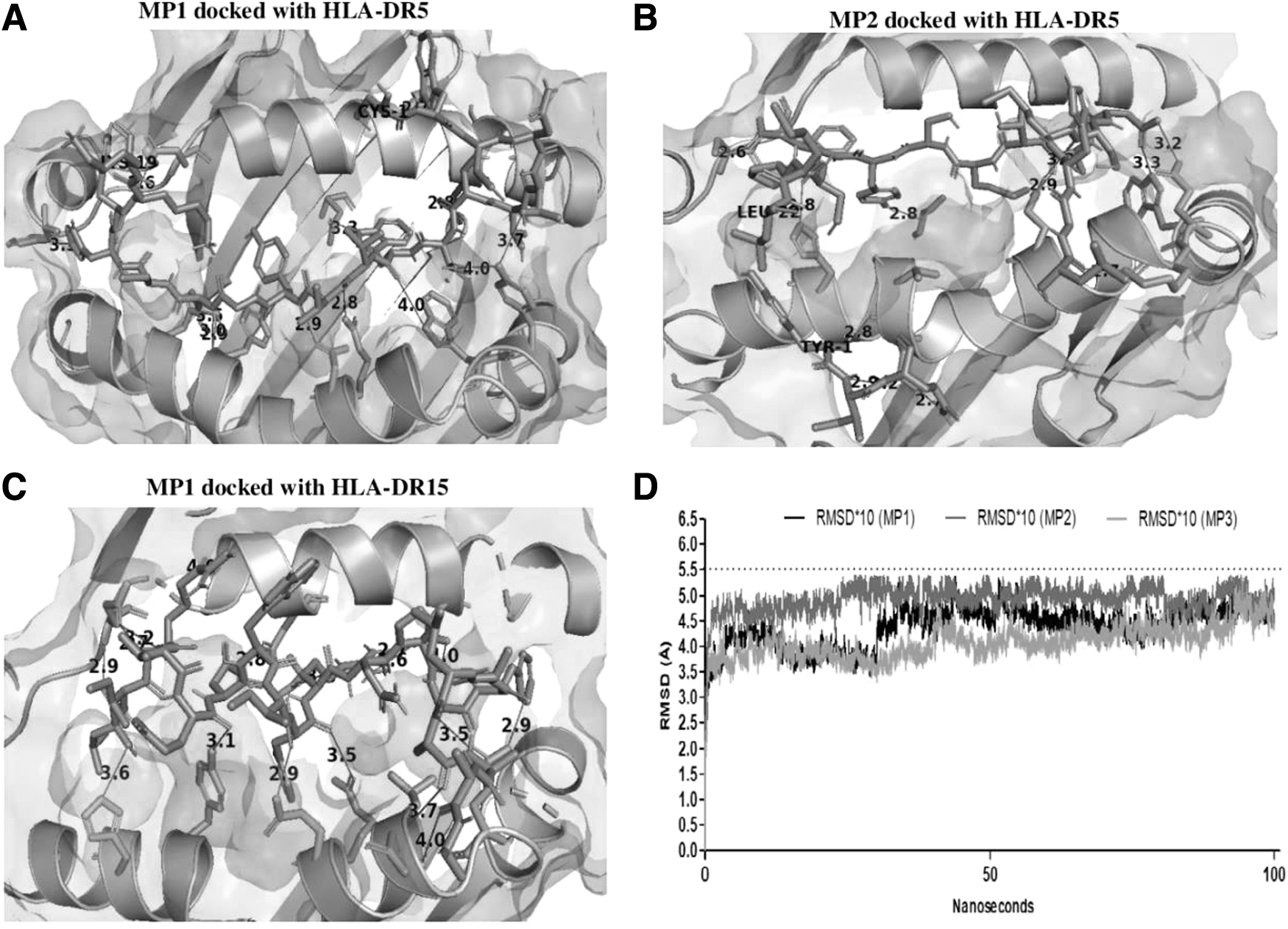
MD simulation
The stability of the best docked pHLA complex due to the biophysical interactions with surroundings was investigated by MD simulations.
All MD analyses considered complete HLA molecules contacted with peptides at the period of 100 ns within the explicit water box. The complexes were solvated with the incorporation of Na+ and Cl− ions. The steepest descent energy minimization of pHLA complexes was done at 5,000 steps. Two-step equilibrations at 2000 ps were carried out for eliminating the steric clashes of amino acids of HLA molecules and peptides. The density, temperature, and pressure of each system were distributed evenly in the equilibrium phase. The full simulation was done with 100 ps for all the subjected pHLA complexes at 300K. The potential energies (PE) were reported as −2.50e+05, −2.28e+05, and −2.01e+05 for E2, E7, and E9 pHLA complexes, respectively. Structural deviations of pHLA are reported through RMSD plots in Figure 6D. It was observed that all three complexes achieved constancy from the beginning of MD but pHLA (E7:HLA-A24) complex showed RMSD fluctuation after nearly 50 ns but after that, all showed less magnitude of deviation.
Likewise, MP1, MP2, and MP3 peptides secured −1.08e+06, −1.56e+06, and −1.05e+06 PE, and their RMSD values are displayed in Figure 7D. It can be seen from the graph that all the complexes obtained similar RMSD values, which remained steady throughout the period. According to the literature (Yang et al., 2014) for the stability of the protein complex, RMSD values around 5.1 Å are considered appropriate. The RMSD values of all selected complexes did not exceed 5.5 Å throughout the simulation course, indicating the stability of the pHLA complexes.
Structure modeling and marking the peptides
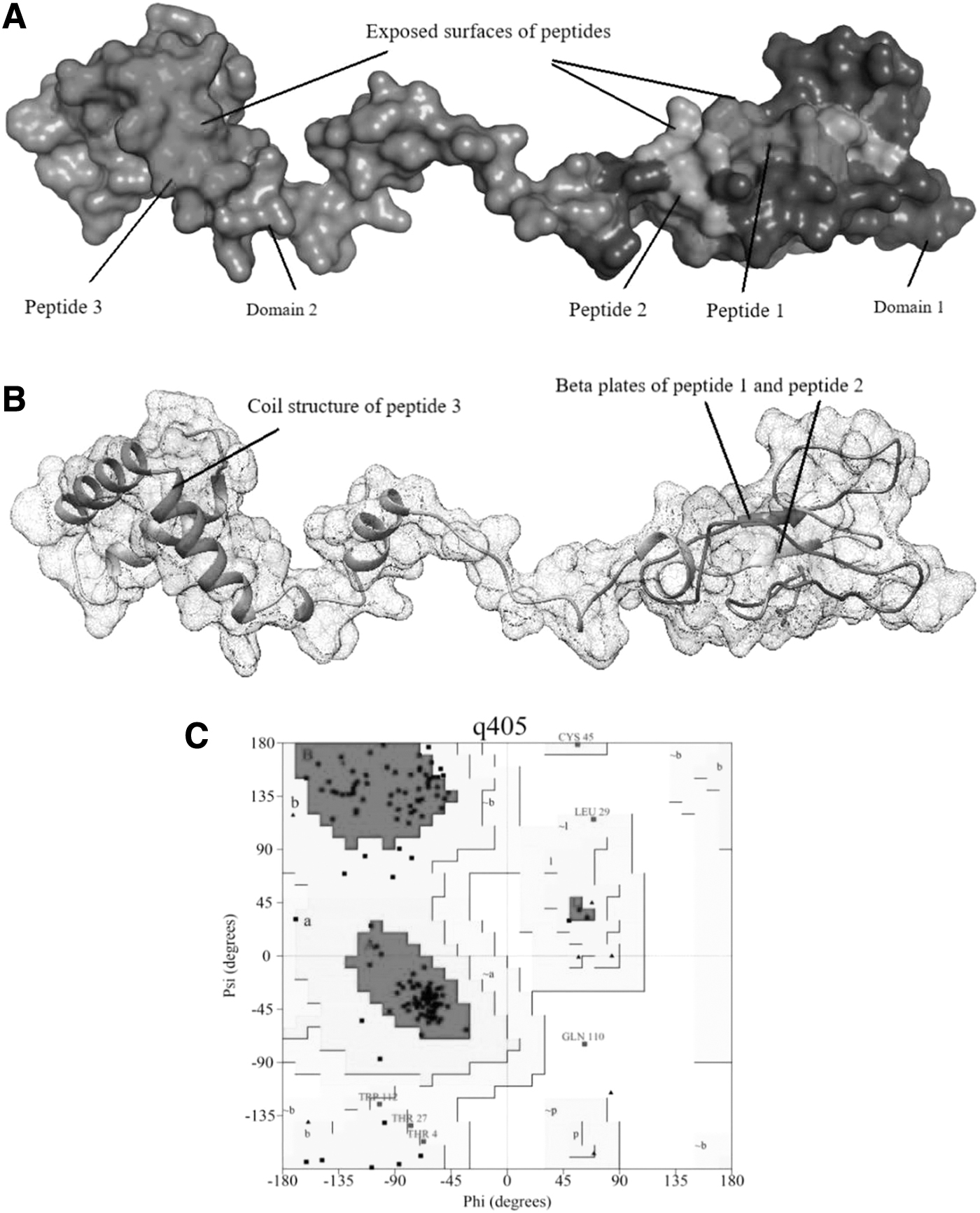
The three-dimensional structure of prM protein was formed by the Modeller tool through homology modeling. The conclusive peptides were marked within the 3D structure of the prM protein, shown in Figure 8A. The refined model was obtained by minimizing the model energy from the initial (38,180.936 kJ/mol) to the final (−6,963.183 kJ/mol) through Chimera tool using 100 steepest descent steps with 10 conjugate gradient steps. The Z-score of the model was found as −4.83 through the ProSA server. The Ramachandran plot of the developed structure, shown in Figure 8C, displaying 84.1% residues occurred in most favored regions, 11.9% residues appeared in additional allowed regions, 3.3% and 0.7% residues existed in generously allowed regions and disallowed regions, respectively. The secondary structure of the prM protein is shown in Figure 8B, which revealed the peptide positions. Peptides were composed of β-plated sheet and α coil structures with exposed surfaces, which revealed their good binding probability.
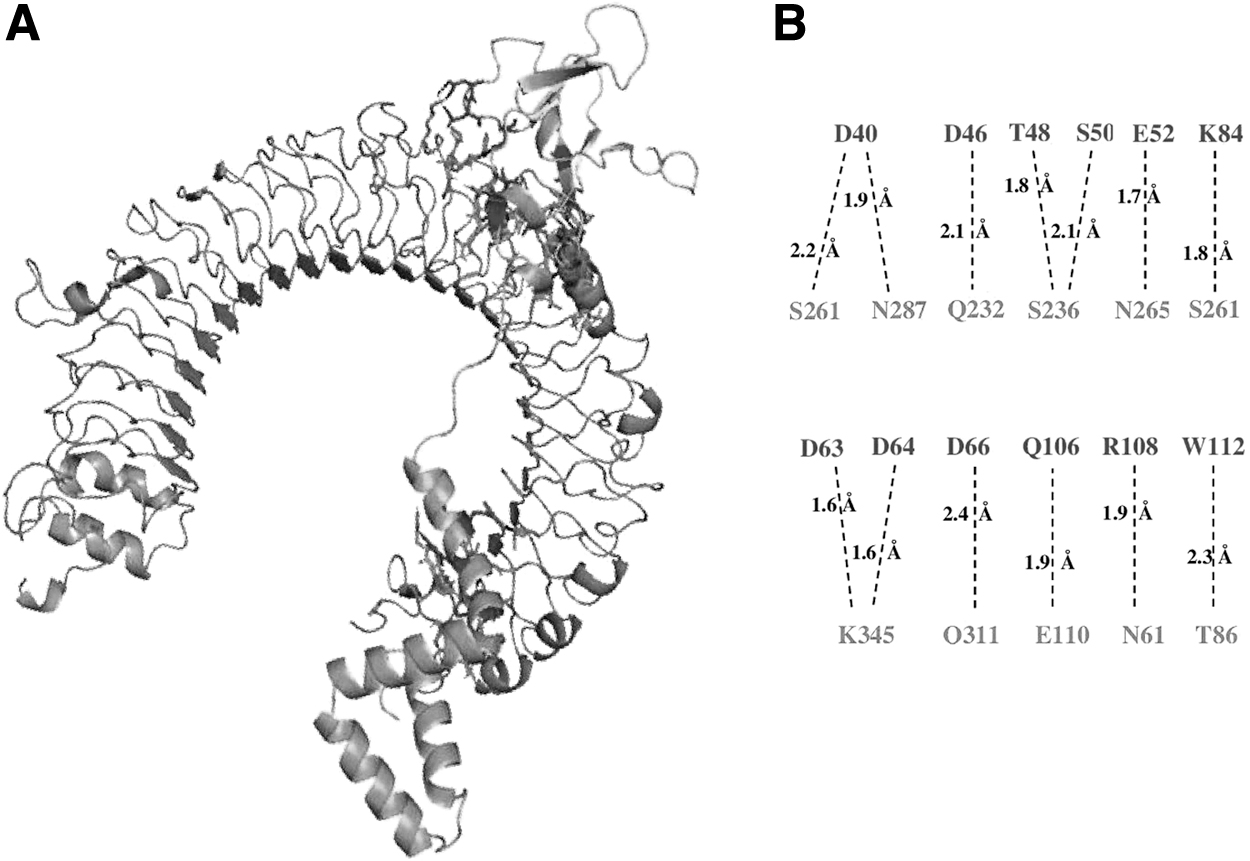
Docking of prM protein with immune receptor
An effective immune reaction is induced through interactions between pattern recognition receptor and antigen. Molecular docking was performed with the HAADOCK server to confirm the interactions between TLR3 and prM protein. The prM protein model reported in Figure 8A was used to dock with TLR3. The residues present in the prM protein (D40, D46, T48, S50, E52, D63, D64, D66, K84, Q106, R108, and W112) participated in the hydrogen bonding as shown in Figure 9. The prM protein showed good binding with TLR3 receptor with good HADDOCK score, −103.6 and 0.7 Å RMSD from the overall lowest-energy structure. The protein (prM) showed strong affinity toward the immune receptor (TLR3) due to strong hydrogen bonds having bond length <3.0 Å clarified through the result.
Analysis of previous report of selected peptides
Selected final peptides (MP1, MP2, and MP3) of Zika prM protein was found similar to peptide fragments reported in IEDB data resource (Table 5). Partial sequence, CNTTSTWVVYGTCHH (related to peptide MP1), was found positive in human B cell assay (Falconi-Agapito et al., 2021). Two partial sequences, QIMDLGHMCDATMSY and LGHMCDATMSYECPM (MP2 peptide) also showed positive B cell response (Falconi-Agapito et al., 2021). Similarly, partial sequences (MP3 peptide) IRVENWIFRNPGFAL, NWIFRNPGFALAAAA, LIRVENWIFRNPGFA, VENWIFRNPGFALAA, and ENWIFRNPGFALAAAA showed positive B cell response (Falconi-Agapito et al., 2021), and IFRNPGFALAAAAIA showed positive T cell response (Hassert et al., 2018).
Previously Reported Sequences of Identified Peptide Fragments
Discussion
Fetal microcephaly and congenital abnormalities are very devastating and vicious human diseases. Several congenital neurological abnormalities have been found in newborns contracting the Zika virus, mainly neurological disorders (Radaelli et al., 2020; Santana et al., 2021). Dependence of Zika flavivirus on Aedes mosquito host and persistent virus outbreaks can be very deadly for future generations. Paixao and his team observed a 52.6 mortality rate for surviving children with Congenital Zika Syndrome in Brazil during the 2015–2018 Zika outbreak (Paixao et al., 2022), which was related to underdeveloped brain and nervous system.
Various vaccines against the Zika virus are in the clinical trials phase I/II, including nucleic acid (DNA, m-RNA)-based vaccines, inactivated whole virus and live attenuated viral vectored vaccines (Pattnaik et al., 2020). These vaccines are safe but require boosters for prolonged protection. In one study, the synthetic DNA vaccines (GLS-5700) encoding Zika prM-E antigen, showed protection against the Zika virus in humans after inducing binding antibodies in all participants, and 62% of participants developed neutralizing antibodies (Tebas et al., 2021). This study also showed that postvaccination sera protect 92% of Zika challenged IFNAR (interferon-α/β receptor) knockout mice. In contrast, another DNA vaccine based on NS1 antigen failed to confer the positive response (Grubor-Bauk et al., 2019). Also, live attenuated and viral-vector vaccines have important issues with safety considerations, such as having the reverse virulent form of the virus and preexisting immunity to the vector virus, which can result in a harmful unwanted immune response or lack of immune stimulation, respectively, especially in case of pregnant women or fetal harmlessness (Pattnaik et al., 2020).
A flavivirus-based chimeric vaccine, rZIKV/D4Δ30–713, is in phase I clinical trials, but the major problem with this is crossreactive antibodies that can alter the immune response generated.
Peptide-based vaccines showed a new direction for defense against pathogens due to their safe, robust efficacy, and power to strongly activate immune response. This response is mainly carried out by effective multiple epitopes consisting of peptides through stimulating T cells, including CD8+ and CD4+ lymphocytes. Several peptide-based vaccines are in clinical trials for malaria, HIV, Cytomegalovirus, Genital Herpes, Alzheimer's disease, cancer, and influenza virus redressal (Li et al., 2014). Conventionally uncovering the optimal immunogens are difficult because of the prolonged tiresome processes. Virtual screening of potential immunogens through computational tools is an adequate way to minimize the cumbersome traditional process. Several studies have determined suitable multiepitope vaccine candidates from different proteins of the Zika virus based on immunoinformatics approaches (Antonelli et al., 2022; Salvador et al., 2019; Shahid et al., 2020), which explain the robustness of this approach.
In this context, we analyzed the significance of the prM protein of the Zika virus and identified strong immunogenic peptides containing multiple epitopes that activate T cells, which may prove suitable for developing peptide-based vaccines against the Zika virus.
The important activities of the virus life cycle, such as the formation of mature virus particles, protection against acidic degradation of the virus envelope, and the presence of conserved amino acid sequence, explain the importance of the prM protein (Nambala and Su, 2018; Yoshii et al., 2012).
In most developing preventives against the Zika virus, the prM protein has been used as a target (Pardi et al., 2017; Pattnaik et al., 2020). The dataset used in this study included 345 complete nonredundant sequences of prM proteins. The outcomes of the phylogenetic study of prM protein suggest that proteins have not had an influential change in evolution. Even clades related to 2015 to 2016 do not show much difference in amino acids. In a phylogenetic study by Assis et al. (2020), considerable changes in the genome of the Zika virus were not observed across epidemic years and regions. The mutational analysis also showed that most of the amino acid variations are less frequent, which is in agreement with phylogenetic analysis. The robustness of any vaccine on multiple strains of the pathogen and generating an appropriate immune response in the host indicates its success.
The conserved amino acid and mutated position are important for any peptide immunogen to be broadly applicable. Numerous research studies conveyed that peptides with conserved sequences showed a significant immune response to targeted pathogens (Jahangiri et al., 2018; Li et al., 2022). It is observed that the involvement of mutations plays a role in pathogenicity by evading T cell response in the case of HIV. A study in mice showed that this single mutation exhibits significant connectivity to microcephaly and neonatal mortality in mice and is responsible for increased Zika infectivity in NPCs of both humans and mice (Yuan et al., 2017). Our analysis found that the mutation frequency at only a single position 17 was 0.209 in 345 prM protein sequences where the Serine was replaced by Asparagine, except that all mutations were found at different positions below a rate of 0.05. This single amino acid change was found prominently in the circular viral sequences during the 2015 to 2018 outbreak. Studies confirm an association between virus outbreaks and neonatal microcephaly during the same period (Brady et al., 2019; Oliveira et al., 2020).
It was reported that the Flavi_M domain has a membrane-anchored region essential for viral protein entry into the host cell membrane and the binding site, known as the glycoprotein E-binding interface (Lu et al., 2020), and this region is also involved in peptide binding. In this context, two functional domains, Flavi_ProPep and Flavi_M, identified in the prM protein were found to belong to the Flavivirus polyprotein propeptide and Flavivirus envelope glycoprotein M-like superfamily, respectively. The CATH server revealed that the Flavi_M domain associated with the flavivirus envelope glycoprotein M-like superfamily was found to be directly associated with protein-binding molecular activity, which is reported through Gene oncology term (GO: 0005515).
An effective antigen must have epitopes that show strong binding to the HLA through which it is presented. Artificial neural network and SMM algorithm-based computational tools have been reported to identify improved epitope BA for HLAs. NetMHCIpan (Jurtz et al., 2017), NetCTLpan (Stranzl et al., 2010), and NetMHCIIPan (Jensen et al., 2018) servers use naturally eluted ligand and BA datasets to predict potent HLA binders. Several studies applied them in a vaccine design against SARS-CoV-2 (Singh Slathia and Sharma, 2020), Nipah virus (Krishnamoorthy et al., 2020), and Rubulavirus (Siañez-Estrada et al., 2020). We used a consensus approach for epitope prediction by employing three different tools for CD8+ and CD4+ T cells with the maximum number of HLA alleles.
It has been evident that combining CD8+ and CD4+ T cell epitopes can elongate the length and strengthen of the peptides for stable and more immune stimulation (Maslak et al., 2018; Nelde et al., 2021). Peptides that consist of linear B cell epitopes can readily be used for immunizations and antibody production. The suitable surface accessible sequential amino acids were present in peptides MP1, MP2, and MP3, confirming the presence of B cell epitopes.
Three peptides were selected in this study, which contains multiple CD8+ and CD4+ T cell and B cell epitopes.
Some studies have shown that prophylaxis vaccination triggers certain autoimmune reactions in individuals. Researchers reported autoimmune hepatitis development after vaccination in the case of SARS-CoV-2 (Rocco et al., 2021) and influenza virus vaccine (Sasaki et al., 2018). Since safety with efficacy is critical for any immunogen, the BLASTp analysis was done with the human proteome after a conservancy check of sequences to remove autoimmune regions.
Toxicity, antigenicity, and stability are the main concerns in peptide-based vaccine development. Various computational studies are conducted in these regards (Ahmed et al., 2022; Rahman et al., 2020). The selected peptides were found to be the highest antigenic with values of 0.8 (MP1), 1.09 (MP2), and 0.46 (MP3) antigenicity besides being nontoxic and nonallergenic. A study carried out by Badani et al. (2014) showed the importance of the hydrophobicity and propensity of antiviral peptides of enveloped viruses for their interactions with other proteins. Studied isoelectric point, weight, stability, thermostability, and hydropathic properties unveil the physicochemical characteristics that support the selected peptides to be stable and effective antigens.
Analyzing the previous reports of the IEDB epitope database, it was found that exact selected peptide sequences have not been reported. The partial sequence of peptide MP1, MP2, and MP3 responded positively in the human B cell assay (Microarray qualitative binding) and the T cell assay (ELISPOT, IFN-γ release IL-2 release) for CD8+ and CD4+ T cells (Falconi-Agapito et al., 2021; Koblischke et al., 2018; Hassert et al., 2018), which supports the antigenicity of our selected peptides.
This study indicated that peptides are capable of inducing T and B cell immune responses.
Misra et al. (2011) used the population coverage tool to clarify a correlation between population coverage and estimated HLA-epitope restriction that reflects epitope prevalence. It has been reported that population coverage above half percentage is suitable for eliciting a good antigenic response (Fatoba et al., 2021). Our analysis found a good association between epitope-bound HLA alleles and their population coverage. The different HLA genotypic coverage by epitope in all geographic regions suggests that these antigens can activate the T cell immune response in a large proportion of individuals around the world.
Molecular docking is known to be a reliable and accurate mode to analyze the binding of peptides to HLA (Patronov et al., 2011). A current study found reasonable mAbs' (monoclonal antibodies) production in two rabbit models against multiple serotypes of dengue virus using a peptide vaccine candidate that was identified, in silico, and docking interactions as an antigen, and confirmed by ELISA (Kaushik et al., 2022). The main purpose of MD simulation is to analyze the dynamic behavior of proteins caused by interactions with other proteins and surroundings over time (Hollingsworth and Dror, 2018).
Residues of a specific peptide bind to HLA in a specific manner when peptides are bound correctly (Liu and Gao, 2011). Prasasty et al. (2019) found promising T and B cell epitopes of the Zika virus in silico manner and performed docking and MD simulation with the best selected peptides from which they received the effectual peptides. The nonamer peptides (CD8+ T cell epitopes) we predicted were fully engaged with the HLA groove active residues. The residues of CD4+ T cell peptides were fully buried in the binding slot, except for the anchoring residues due to their large length.
All peptides were well docked, of which three peptides from CD8+ T cells achieved good HADDOCK scores, −148.8, −137.8, and −137.6. Similarly, −187.2, −170.5, and −181.2 scores were obtained by CD4+ T cell peptides whose values were higher than those of the docked NP. The RMSD value of pHLA not exceeding 5.5 Å during the entire tenure of the MD simulation indicates their stability.
It has been found that peptides present in the β-sheet or α-helix support the formation of scaffolds and flanking regions due to amino acid propensities that increase the antigenicity of the peptides (Jackson et al., 2002). A 3D model of the prM protein was built to determine the structural conformation of selected peptides within the protein. The Ramachandran plot and Z-score confirmed the correctness of the constructed prM protein model. From the 3D model, we found that the peptides MP1 and MP2 were present in domain 1 (Flavi_ProPep) and MP3 existed in domain 2 (Flavi_M). The peptides were in α-helix and β-plated sheet structural forms that support their antigenicity. The α-helix and β-plated sheets are the secondary structures of protein, which are more stable due to hydrogen bonds between the amide and carboxyl groups. These structures enable protein folding and protein–protein interactions, therefore, facilitate peptides to the stable binding that contributes to their antigenicity (Groß et al., 2016). TLRs are critical in triggering an innate immune response by binding with diverse pathogen molecules (Gao and Li, 2017).
A recent study reports that they also participate in the activation of antigen-presenting cells, antigen loading, and cytokines regulation (Duan et al., 2022). We checked the affinity of prM protein toward the TLR through docking with a good HADDOCK score −103.6 and 0.7 Å RMSD value. This study shows the prominence of prM protein as a potent antigen and provides three peptides (MP1, MP2, and MP3) that are highly antigenic, immunogenic, and nonallergic with good population coverage, containing potential 9 and 13 epitopes for CD8+ and CD4+ T cells, respectively, and 9 B cell epitopes.
Conclusions
Phylogeny, mutational, conserved domain, and human protein interactions' study demonstrated that the prM protein is essential as a definite contender for therapeutic gain. The predicted epitopes and constructed peptides in this study expressed good binding behavior with extensive HLA alleles distributed worldwide. Identified potent T and B cell epitopes through immunoinformatics analysis may aid in the experimental design of effective peptide-based vaccines to address the Zika virus challenge.
Footnotes
Acknowledgment
The authors would like to thank the scientific community.
Authors' Contributions
Y.G.: Draft writing; data curation; analysis; designing. M.B.: Conceptualization; methodology; supervision; and review and editing. B.C.: Visualization; and review and editing.
Consent to Participate
All the coauthors consent to participate and are aware of and approve of the submission.
Data Availability Statement
The protein sequences were retrieved from NCBI databases and all data generated or analyzed during this study are included in this article and its support information.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.