
Abstract
Infectious bronchitis virus (IBV), a gammacoronavirus within the Coronaviridae family, is an economically important etiological disease agent in chickens. Both early diagnosis and determination of the immune status of chickens are important for controlling IBV outbreaks in chicken flocks. The N protein is the most abundantly expressed virus-derived protein during IBV infection and can induce a strong immune response by producing antibodies during early infection or immunization. In this study, we found that the amino acid sequences of the N protein between CK/CH/LJL/04I and the other 22 IBVs were conserved, especially in the 1–160 amino acid region. Based on the sequence similarities, the three recombinant proteins, rN160 (amino acid positions 1–160), rN266 (144–409), and rN409 (1–409), were expressed using the Escherichia coli system and subsequently purified. The results demonstrated that the antigenicity and reactivity of rN160 were better than those of rN266 and rN409. As a result, an indirect enzyme-linked immunosorbent assay (ELISA) (rN160 ELISA) was developed to detect the IBV antibody based on the rN160 protein. Using 1,500 clinical field serum samples, the relative sensitivity, specificity, and accuracy of the rN160 ELISA were 98.97%, 92.34%, and 97.93%, respectively, compared to those of a commercial ELISA kit (IDEXX), indicating a strong positive correlation between the two methods. Taken together, these results reveal that the rN160 ELISA is a rapid, simple, and sensitive method for detecting group-specific IBV antibodies for epidemiological investigation and antibody-level monitoring.
Introduction
Infectious bronchitis virus (IBV) is the agent of an economically important etiological disease, infectious bronchitis (IB), in chickens (Cavanagh, 2005; de Wit and Cook, 2019); IBV affects chickens of all ages and causes a range of clinical signs depending on the virus strain, age, and chicken, including respiratory disease, nephritis, and a reduction in egg production in laying hens (Cavanagh, 2007).
The IBV genome is an unsegmented, single-stranded positive-sense RNA ∼27.6 kb in length (Cook et al., 2012). The 3′-end of the viral genome, which occupies approximately one-third of the viral genome, encodes viral structural and accessory proteins in a sequence of spike (S), accessory proteins 3a and 3b, envelope (E), membrane (M), accessory proteins 5a and 5b, and nucleocapsid (N). The 5′-end, which encompasses approximately two-thirds of the genome, encodes two overlapping replicase polyproteins (1a and 1b), which are further processed into 15 nonstructural proteins (Nsp2–Nsp16) (Lim et al., 2016).
Methods based on the detection of antibodies against IBV are routinely used to investigate the previous circulation of field strains or assess the vaccine-induced immunological response. Enzyme-linked immunosorbent assay (ELISA) is one such method that has been developed and proven to be efficacious (Legnardi et al., 2020). Since its first report in 1981, ELISAs using inactivated IBV particles as the coating antigen have been used worldwide to evaluate the level of IBV-specific antibodies (Marquardt et al., 1981).
At present, inactivated whole virus is the most commonly used coating antigen in the commercially available diagnosis kits used for IBV diagnosis (Cook et al., 2012; Legnardi et al., 2020). Nevertheless, whole IBV particles are tedious to produce, requiring virus proliferation and purification, resulting in a high cost of commercial ELISA kits, which limits their application. Many studies have indicated that recombinant S and N proteins from prokaryotic, yeast, or baculovirus systems as coating antigens in ELISA could provide a rapid and large-scale method for the detection of IBV antibodies (Chen et al., 2003; Gibertoni et al., 2005; Gu et al., 2022; Lei et al., 2017; Lin et al., 2012; Ndifuna et al., 1998).
The S protein of IBV showed high diversity among IBV strains, which restricted its application for detecting IBV group-specific antibodies (Promkuntod et al., 2014; Shang et al., 2018; Wickramasinghe et al., 2014). However, the phosphorylated N protein is a conserved structural protein with a high degree of identity (91.0–96.5%) among different IBV strains (Niesters et al., 1986), as well as being the most abundantly expressed virus-derived protein during IBV infection, which can induce a strong immune response for producing antibodies, especially in early infection or immunization (Cavanagh, 2005; Chen et al., 2005; Zhang et al., 2021). These advantages make IBV N an ideal target for diagnosis, both by serological and molecular methods (Chen et al., 2003; Cook et al., 2012; Lugovskaya et al., 2006).
Recent studies have shown that the N protein has varying degrees of variation among IBV strains, some of which are irrelevant to antibody production (Park et al., 2005; Sapats et al., 1996). To this end, we analyzed the published sequences of the N gene in the GenBank database from various IBV strains, representing different genotypes and lineages. Based on the epitope analysis of the N protein, three recombinant N proteins, including rN160 (amino acid positions 1–160), rN266 (144–409), and rN409 (1–409), were expressed using the Escherichia coli system, and then purified. rN160 showed advantages regarding the expression amounts, antigenicity, and reactivity compared to those of other two recombinant proteins. As a result, an indirect ELISA (rN160 ELISA) was developed and evaluated to detect the IBV antibody based on the rN160 protein.
Materials and Methods
Viruses
An IBV strain, CK/CH/LJL/04I (accession number: AY839143), was selected and used to amplify and clone the N gene. The virus was isolated in 2004 from the swollen proventriculus tissues of infected layer hens in Jilin Province, China (Liu et al., 2006). CK/CH/LJL/04I, a QX-like IBV strain, belongs to the GI-19 lineage based on the S1 gene full-length sequences. GI-19 is the predominant IBV type in China and is one of the most important IBV genotypes globally (Valastro et al., 2016).
Chickens
Specific pathogen-free (SPF) chickens from Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences, were used in this research. The Ethical and Animal Welfare Committee of Heilongjiang province, China, provided ethical approval to conduct this study (License no. HSY-IACUC-2019-129).
Sera
Reference negative sera were collected from SPF chickens of different ages and were confirmed using a commercial ELISA kit (FlockChek1 Infectious Bronchitis Virus Antibody Test Kit; IDEXX, Westbrook, ME). Positive sera against IBV strains 4/91, H120, M41, QX, and other avian viruses, including those of avian influenza virus (AIV), infectious bursal disease virus (IBDV), and Newcastle disease virus (NDV), were also used in this study (Chen et al., 2015; Han et al., 2017; Liu et al., 2013).
To prepare serum samples from chickens vaccinated with the IB vaccine, 24 SPF chickens were vaccinated through the ocular and nasal routes with 103.5EID50/per bird of the IBV H120 vaccine strain at 15 days of age. Twenty-four uninfected control birds were administered phosphate-buffered saline (PBS). At 4, 8, 12, 16, 24, 32, 40, and 48 days postinfection, three chickens from each group were randomly chosen to be bled to collect sera.
To prepare the reference serum positive for rN160, 3-month-old SPF chickens were immunized subcutaneously with an emulsion mixture containing an equal volume of complete Freund's adjuvant and the purified rN160 (1 mg/chicken). The purified rN160 protein was prepared as described in section 2.4. Two booster immunizations with the same dose of antigen emulsified with equal volume of incomplete Freund's adjuvant were administered to the chickens at 3-week intervals. Fifteen days after the last immunization, sera from different immunized chickens were collected and pooled as positive serum.
One thousand and five hundred clinical field serum samples were obtained from commercial chicken flocks in different provinces of China. Of the 1,500 clinical serum samples, 500 were collected from chickens with an unknown background of IB vaccination, and the remaining 1,000 were from chickens immunized with vaccine strains of 4/91, H120, or LDT3-A (Table 1).
Backgrounds of 1,500 Clinical Serum Samples Collected and Used in This Study
IB, infectious bronchitis.
Sequence alignment
According to the IBV genotype classification (Valastro et al., 2016), a total of 22 sequences of the N gene from representatives of different genotypes and lineages of IBV strains were downloaded from the GenBank database, together with the accession numbers (Table 2). The deduced amino acid similarities of N sequences were calculated between the IBV strain CK/CH/LJL/04I, and the 22 other IBV strains. The amino acid sequences were then analyzed using MegAlign from the DNAstar software package (DNASTAR, Inc., Madison, WI).
Backgrounds of Representative Infectious Bronchitis Virus Strains with Available N Gene Sequences Used in This Study
The N gene sequences of GI-10, GI-11, GI-12, GI-20, GI-12, GI-23, GI-25, GI-26, GI-27, and GIV-1 were not available in GenBank database and had not been included in this study.
IBV, Infectious bronchitis virus.
Cloning and expression of the N160, N266, and N409 genes/fragments
The viral RNA was extracted from allantoic fluids infected with the IBV strain CK/CH/LJL/04 using TRIzol (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. The N gene was amplified using the PrimeScript™ One-Step RT-PCR Kit (Takara, Beijing, China) (Liu et al., 2013). The primer sets were designed according to the published sequence of the IBV N gene CK/CH/LJL/04. Forward primer (rN160F: 5′-CGCGGATCCATGGCGAGCGGTAAAGCAAC-3′) and reverse primer (rN160R: 5′-GCGTCGACTTACAGAGGAATAAAATCCCAACGG-3′) were used to amplify N160. The forward primer (rN266F: 5′-CGCGGATCCATGTCAGATGGAGGACCTGACGG -3′) and reverse primer (rn266rs: 5′-GCGTCGACTTATCAAAGTTCATTTTCACCAAG-3′) were used to amplify N266, and rN160R and rN266R were used to amplify N409.
Each amplified fragment was then cloned into the cloning vector pMD18-T for sequencing. Then the fragments of N160, N266, and N409 were digested with the restriction enzyme (BamH I and Sal I) and ligated into the expression vector pGEX-6p-1 with T4 DNA Ligase (Takara, China). The directionality of recombinant plasmids was verified by sequencing. Each plasmid was transformed into E. coli (Rosetta) competent cells. Protein expression was induced by 0.8 mM isopropyl-beta-
Recombinant proteins were expressed with a glutathione S-transferase (GST) tag at the C-terminus, which allowed for further purification by affinity chromatography according to the GST-binding resin manufacturer's protocol. Each of the soluble fractions, which contained the most rN160, rN266, and rN409 proteins, was purified using GST Bind resin (MERCK, Germany), before determining the protein concentration using a spectrophotometer.
Western blotting
Purified rN160, rN266, and rN409 proteins were separated by 12% sodium dodecyl-sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes using a minitrans-blot system (Bio-Rad), according to the manufacturer's instructions. Nonspecific binding to the membranes was blocked with 5% skim milk in PBS containing 0.05% Tween-20 (PBST) overnight at 4°C with gentle agitation. The membranes were then incubated with positive serum against IBV (1:100) at 37°C for 1 h, followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-chicken IgG (1:5,000 dilutions in PBS, pH 7.4) at 37°C for 1 h. Following three washes with PBST, the rN160, rN266, and rN409 proteins were detected using 3,3-diaminobenzidine tetrahydrochloride, which was stopped by rinsing the membrane in deionized water, followed by drying of the membrane.
Detection of IBV antibody using ELISA with rN160, rN266, and rN409 as coating antigens
The rN160, rN266, and rN409 proteins (500 ng/well) diluted in 0.1 M carbonate buffer (pH 9.6) were used as coating antigens in 96-well plates (Nunc Maxisorp, San Diego, CA) and incubated overnight at 4°C. Plates were washed three times with PBST (PBS with 0.05% Tween-20) and then blocked with 100 μL/well 1% (w/v) bovine serum albumin in PBST by incubation at 37°C for 2 h. After washing three times with PBST, specific antibodies against IBV strains 4/91, H120, M41, and QX, and other virus strains, including AIV, IBDV, and NDV, were tested to determine antigenicity and cross-reactivity.
The plates were washed three times with PBST again before adding 100 μL/well of the secondary antibody (goat anti-chicken IgG [H+L], Thermo) and incubated at 37°C for 1 h. The plates were washed again three times with PBST followed by the addition of 100 μL/well of TMB (Sure Blue TMB Microwell Peroxidase; KPL). The plates were incubated for 5 min in the dark, and the reaction was terminated using 100 μL/well stop solution (Blue stop solution; KPL). The plates were read at optical density (OD)650nm using a microplate reader (Bio-Rad, Japan). All tests were conducted in triplicate. The methods using rN160, rN266, and rN409 as coating antigens were named rN160 ELISA, rN266 ELISA, and rN409 ELISA, respectively.
Optimization of the rN160 ELISA
The rN160 protein gave the highest ratio of OD650nm values between the positive and negative sera (P/N value) of the different IBV strains, thereby further optimizing the ELISA. A checkerboard titration was conducted using different coating antigen concentrations of rN160 protein (1.25, 2.5, 5, 7.5, 10, and 15 ng/μL), serum dilutions (1:100, 1:200, 1:500, and 1:1,000), and secondary HRP-conjugated antibody dilutions (1:1,000, 1:2,000, 1:5,000, and 1:10,000). The other assay parameters were optimized, including incubation time for serum samples, HRP-conjugated antibody, and reaction time for the chromogenic substrate. The conditions that gave the highest P/N values were scored under optimal working conditions.
Determination of the cutoff value for the rN160 ELISA
The mean OD of each test sample was expressed as a test sample to positive sample ratio (S/P) in relation to the mean OD650nm of the reference positive serum and the reference negative sera according to the formula S/P = (sample OD650nm–negative control OD650nm)/(positive control OD650 nm–negative control OD650nm). The cutoff value for the optimized rN160 ELISA corresponded to three standard deviations (SDs) above the mean S/P values for 79 negative sera collected from chickens that were known to be free of IBV infection.
Comparison of the rN160 ELISA and commercial ELISA kits
A total of 1,500 clinical serum samples were tested using our rN160 ELISA and the commercial ELISA kit (IDEXX). The commercial ELISA was conducted according to the manufacturer's instructions, and each sample was tested three times. The rN160 ELISA was performed according to the above-mentioned optimal working conditions. The sensitivity and specificity of the rN160 ELISA and commercial ELISA kit were compared using clinical serum samples.
Detection of IBV antibody from chickens vaccinated with H120
IBV antibody was detected in the serum samples using both the rN160 ELISA and commercial ELISA kits (IDEXX).
Statistical analysis
Each experiment was independently replicated at least three times. GraphPad Prism (GraphPad Software, San Diego, CA) was used to analyze the data. Statistical differences were evaluated using one-way analysis of variance (ANOVA) and Tukey's test. All presented data were shown as the mean ± SD using the SPSS software, and differences were considered statistically significant when p-values <0.05.
Results
Sequence analysis of the N protein
The deduced amino acid sequences of N genes were compared between the IBV strain CK/CH/LJL/04I and 22 other IBV strains, representing different genotypes and lineages according to the IBV classification (Valastro et al., 2016). The amino acid similarities of the N gene ranged from 88.75% to 95.84% between CK/CH/LJL/04I and the other 22 IBV strains. CK/CH/LJL/04I shared 89.38–97.5% and 87.15–97.19% deduced amino acid similarities with the other 22 IBV strains between amino acid residues 1–160 and 161–409, respectively, indicating that the N gene sequences between CK/CH/LJL/04I and the other 22 IBV strains were conserved, especially in the 1–160 amino acid region (Fig. 1).
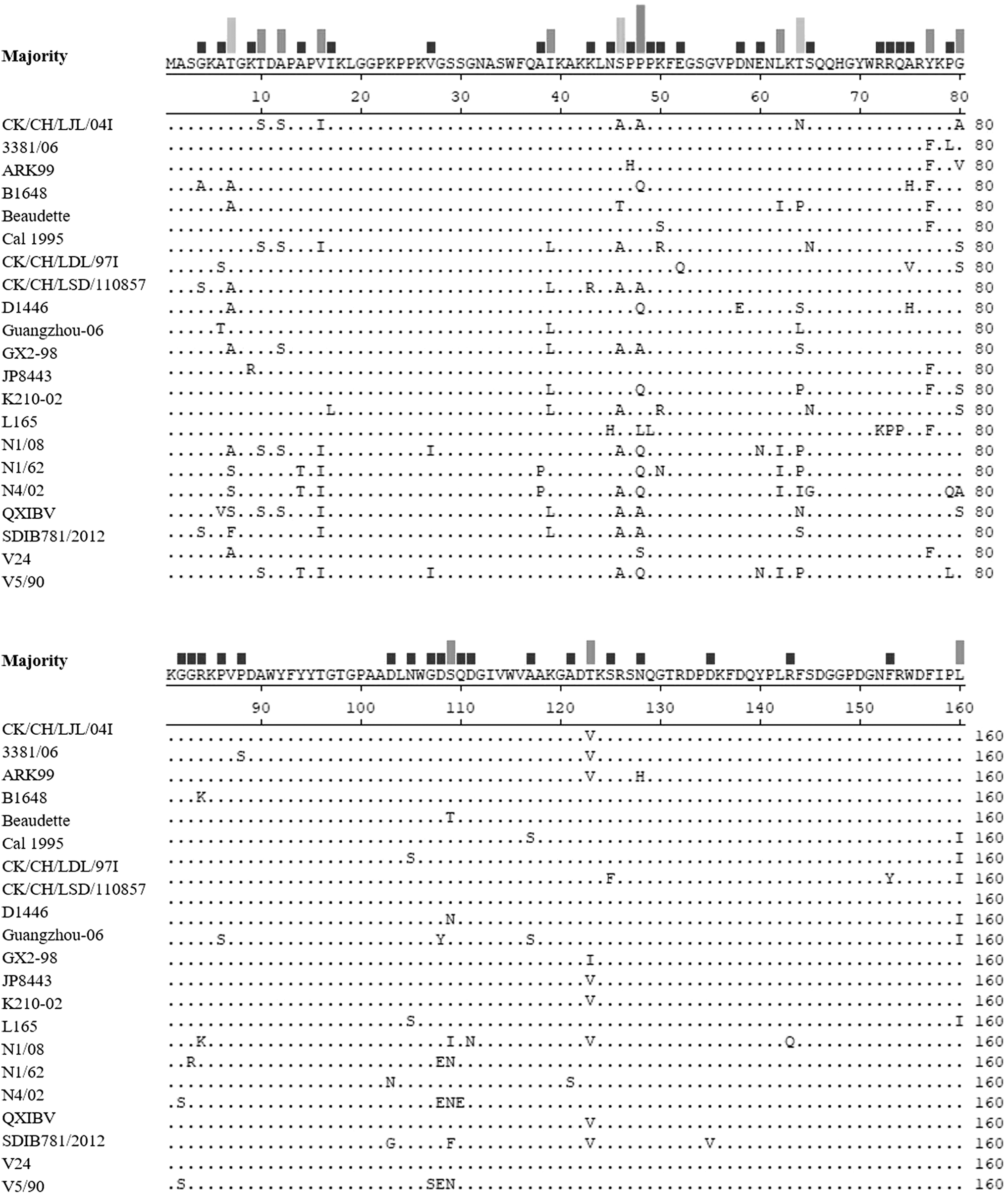
Alignment of the sequence at the 1–160 amino acid region in the IBV N protein. Amino acid residues 1–160 of CK/CH/LJL/04I were aligned with those of the other 22 IBV strains. The dots indicate the regions in which the sequences are identical to the consensus sequences. The deletions within the sequences are indicated by dashes. IBV, infectious bronchitis virus.
Expression, purification, and identification of rN160, rN266, and rN409
The rN160, rN266, and rN409 proteins were expressed in the supernatant after ultrasonic lysis (Fig. 2A–C). The purified rN160, rN266, and rN409 proteins had approximate molecular weights of 43, 55, and 70 kDa, respectively (Fig. 2A–C). Western blotting was conducted using serum positive for IBV to detect the recombinant proteins at their expected molecular weights (Fig. 2D–F). These results indicated that the three recombinant proteins, rN160, rN266, and rN409, specifically reacted with the antibody against IBV in this study.
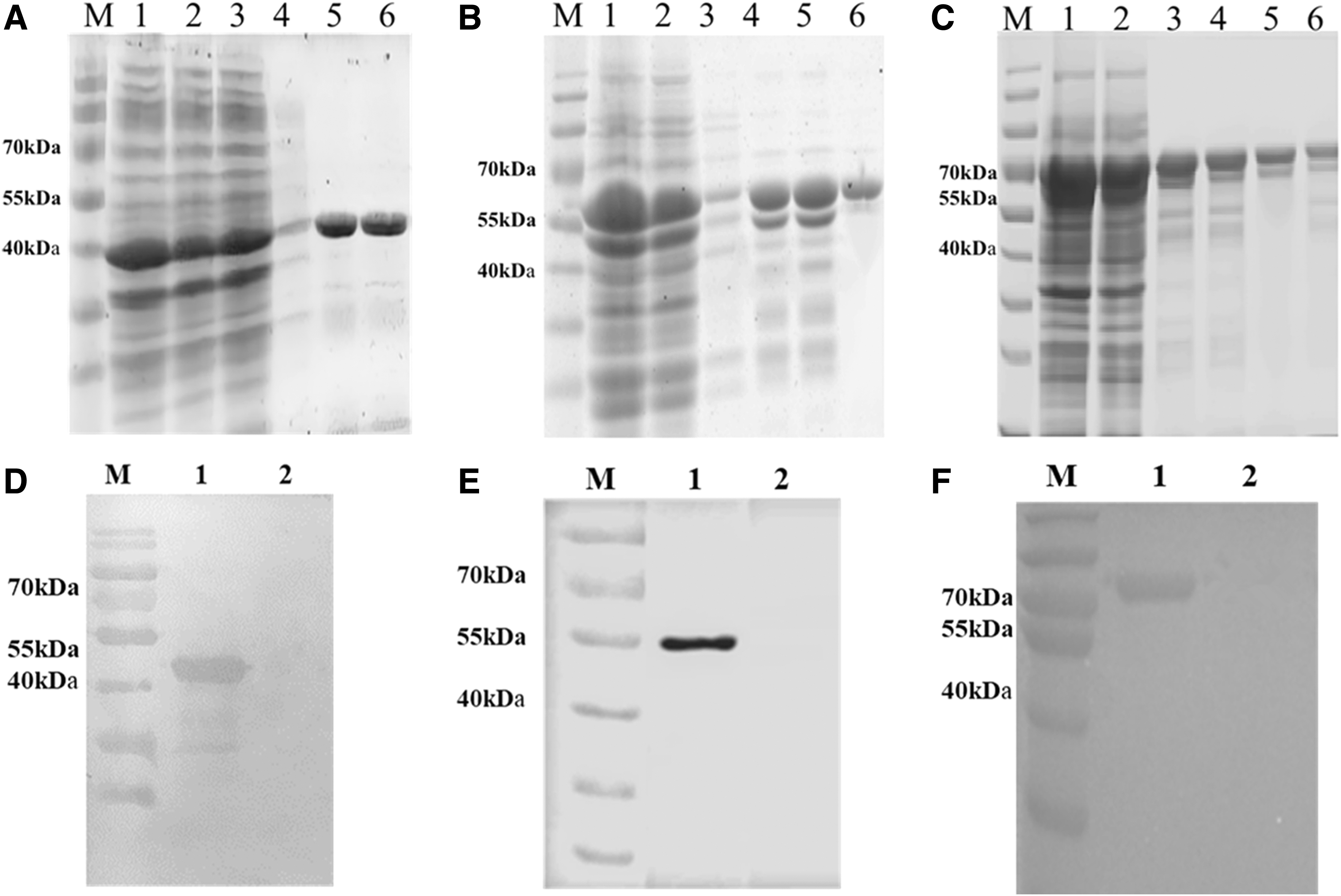
Expression, purification, and identification of rN160, rN266, and rN409 proteins. Identification and purification of rN160
The three ELISAs using rN160, rN266, and rN409 proteins as coating antigens achieved positive values against IBV 4/91, H120, M41, and QXIBV antibodies and negative values against AIV, IBDV, and NDV antibodies (Fig. 3A), indicating that the three ELISAs could distinguish IBV from other chicken pathogens without false positive results or cross-reactivity between different genotypes of IBV strains. Comparatively, rN160 ELISA showed higher P/N values using IBV 4/91-, H120-, M41-, and QXIBV-positive sera than those of rN266 ELISA and rN409 ELISA (Fig. 3B). These data indicate that rN160 ELISA has better reactivity and sensitivity, but has a lower background than those of rN266 and rN409 ELISA. Thus, we chose rN160 ELISA in the following research.

Specificity and sensitivity of rN160, rN266, and rN409 ELISAs.
Optimization of the rN160 ELISA
The results showed that the optimum rN160 protein coating concentration was 5 ng/μL and the optimal dilution of the test set was 1:500. The optimal serum sample incubation time was 45 min, the HRP-conjugated antibody incubation time was 30 min, and the chromogenic substrate reaction time was 5 min (Fig. 4).
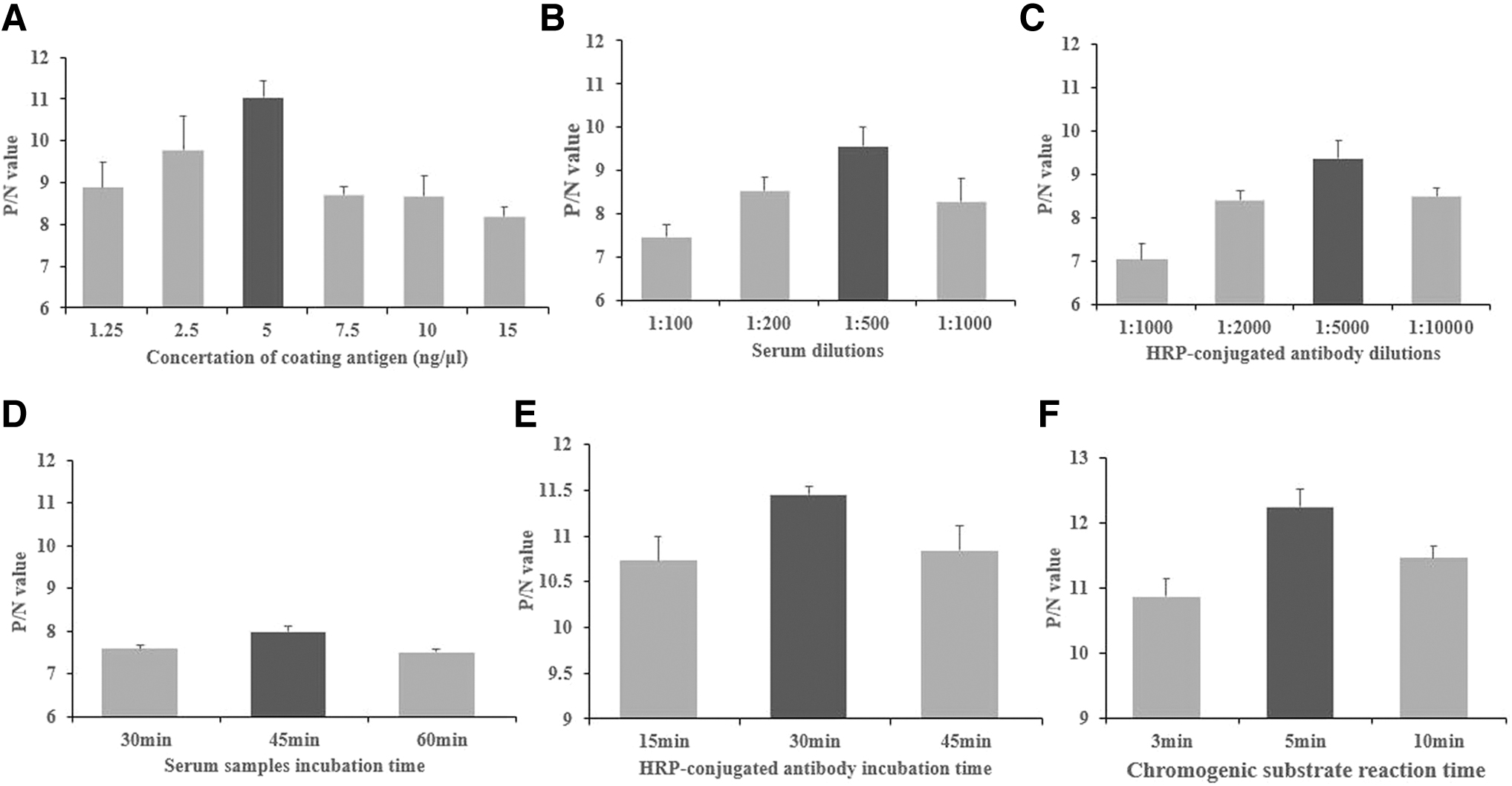
Optimization of the rN160 ELISA system.
After optimization of the rN160 ELISA system, our standardized rN160 ELISA procedure was 100 μL of 5 ng/μL purified N160 protein coated onto the wells of an ELISA plate. After washing, 1:500 diluted IBV-positive and IBV-negative sera were added for a 45-min incubation at room temperature. An aliquot (100 μL/well) of the 1:5,000 diluted HRP-conjugate antibody was added and incubated for 30 min at room temperature. The reactions were stopped by adding 100 μL KPL Blue stop solution after a 5-min incubation with 100 μL KPL Sure Blue TMB Microwell Peroxidase, before reading the plate on a microplate reader (Bio-Rad, Japan) at 650 nm.
Determination of the rN160 ELISA cutoff
The results using 79 serum samples from SPF chickens indicated that the average S/P value of negative serum samples was 0.058, with an SD of 0.046. The cutoff value for the optimized rN160 ELISA, which corresponded to three SDs above the mean S/P values, was 0.2 (Fig. 5). In other words, when the S/P value of the tested chicken serum was ≥0.2, it was determined as the IBV-seropositive sample, and vice versa.
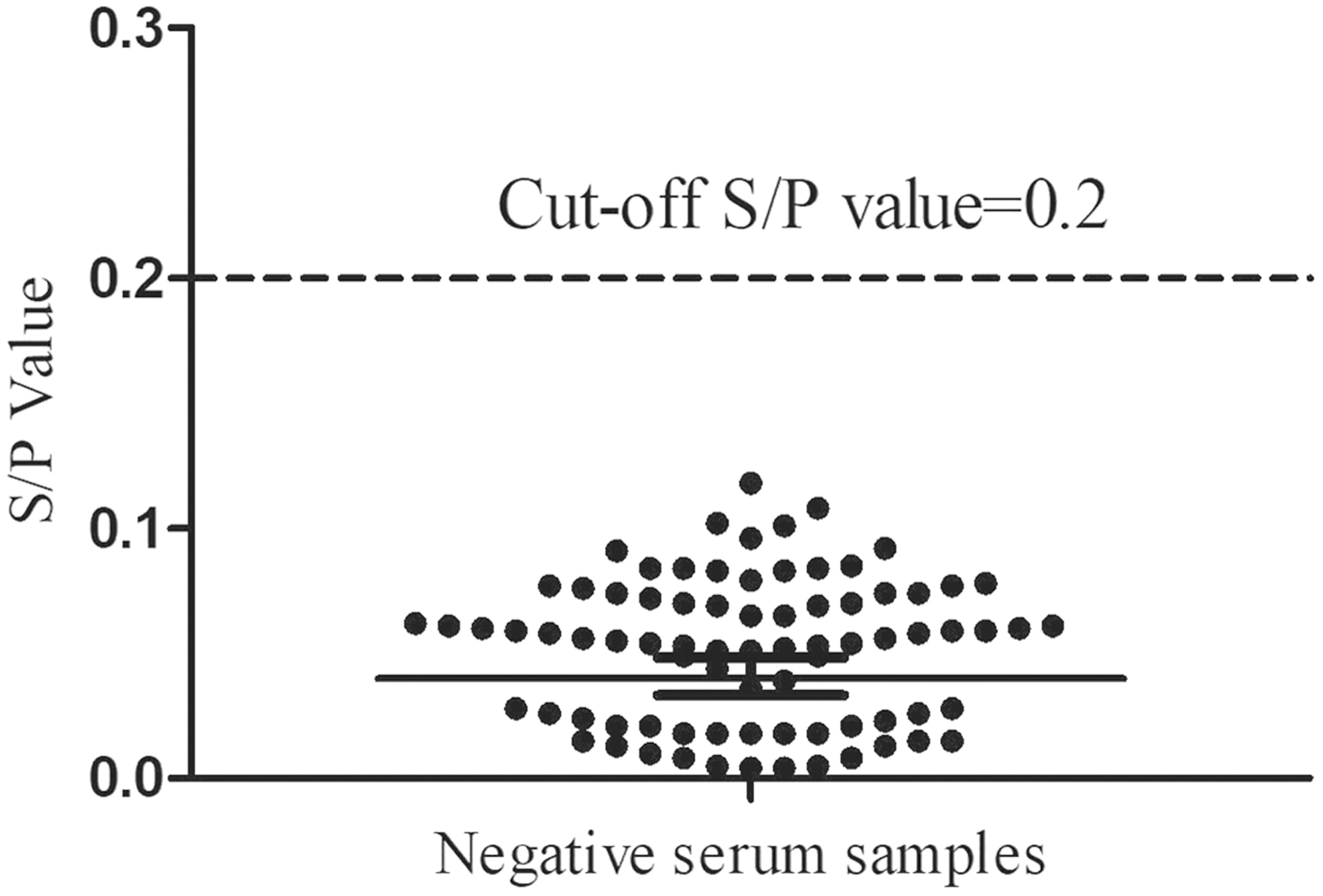
Seventy-nine negative serum samples against IBV were tested using the rN160 ELISA to calculate the cutoff S/P-value.
Comparison of the rN160 ELISA and commercial ELISA kits
A comparison of the rN160 ELISA and commercial ELISA kits was conducted to analyze the sensitivity and specificity of each method. Of the 1,500 clinical field serum samples, 1,265 were determined to be positive and 235 were deemed negative by the commercial ELISA kit. In contrast, the rN160 ELISA determined 1,270 serum samples to be positive and 230 to be negative. Furthermore, 1,252 and 217 serum samples were judged to be positive and negative, respectively, using both methods in unison.
Thirty-one samples gave discordant results, of which 13 were positive only by rN160 ELISA and had low S/P values, although still above the cutoff point. Eighteen were positive only in the commercial ELISA kit. Using the commercial ELISA kit as a reference, the specificity and sensitivity of the rN160 ELISA were calculated to be 92.34% and 98.97%, respectively. The concordance between the two methods was 97.93% (Table 3). Hence, the correlation between the rN160 ELISA and the commercial ELISA kit was both high and significant (p < 0.0001) (Fig. 6).
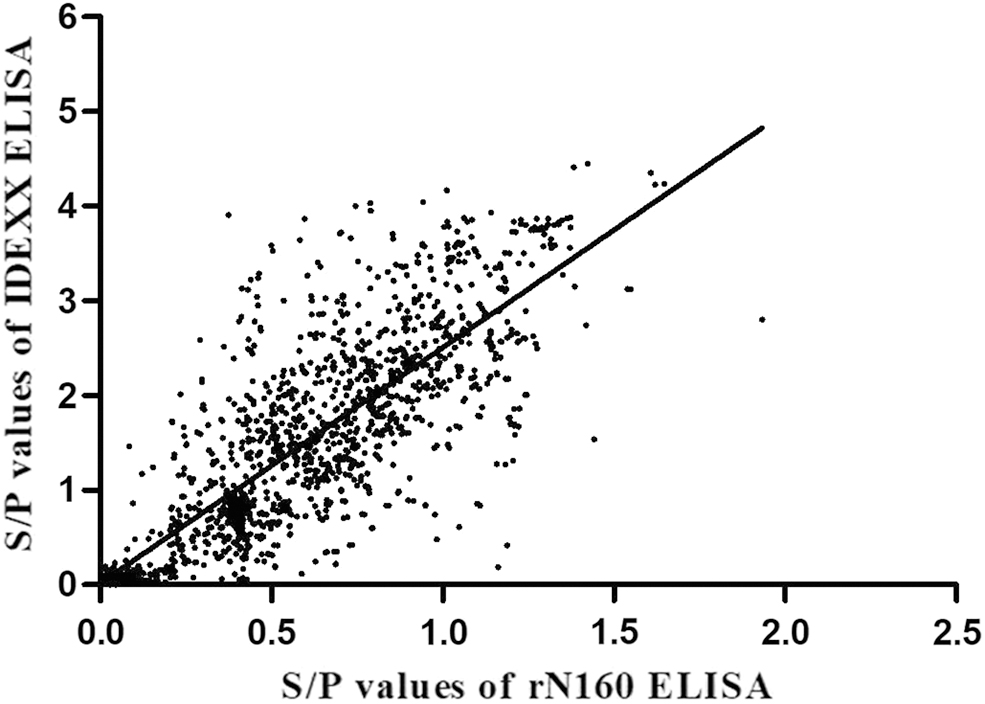
Scatter plot of the S/P-values obtained from 1,500 clinical serum samples using rN160 ELISA and commercial ELISA kits. A strong correlation was observed between the two ELISAs. The formula was as follows: y = 2.4892x + 0.016, r 2 = 0.6584, n = 1,500, p < 0.0001).
Detection of Infectious Bronchitis Virus Antibodies in 1,500 Clinical Sera by rN160 Enzyme-Linked Immunosorbent Assay (ELISA) and Commercial ELISA Kit
Detection of serum samples from chickens vaccinated with the IB vaccine
The rN160 ELISA and commercial ELISA kits were used to measure the IBV antibody in serum samples taken from chickens vaccinated with the IBV H120 strain and control chickens. The chickens were found to be seraconversed at 8 days postvaccination and lasted for the entire experimental period (48 days). No IBV antibody was detected in the control group. The concordance between the rN160 ELISA and commercial ELISA kits was 100% (Table 4).
Detection of Infectious Bronchitis Virus Antibody in Experimental Chicken Serum Samples Vaccinated with Infectious Bronchitis H120 Vaccine
Days postinfection.
Positive numbers/numbers of tested.
Discussion
IBV is an economically important etiological disease agent in chickens (Cavanagh, 2005), which affects chickens of all ages and causes a range of clinical signs, including respiratory disease, nephritis, and a reduction in egg production in laying hens, depending on the virus strain, age, and type of chicken (Cavanagh, 2007). Rapid and sensitive detection methods are essential for epidemiological investigations and for assessing the effectiveness of vaccination. ELISA is the first choice for large-scale disease outbreak monitoring. In this study, we developed an indirect ELISA based on an antigenic region (amino acid positions 1–160) within the nucleocapsid protein for IBV antibody detection.
The antigenicity of the coating antigen is a crucial factor when performing serological tests such as ELISA for antibody detection (Chen et al., 2003; Cook et al., 2012; Lugovskaya et al., 2006). The N protein is highly conserved among IBV strains and has been shown to be capable of inducing a strong immune response for producing antibodies, especially in early infection or immunization (Cavanagh, 2005; Chen et al., 2005; Zhang et al., 2021). In this study, high deduced amino acid sequence similarities were found between the N genes of CK/CH/LJL/04I and other IBV strains, especially in the 1–160 amino acid region, indicating that this region in the N gene is suitable as a target for IBV diagnosis.
The rN160 ELISA described in this study showed better reactivity and sensitivity and a lower background compared with the rN266 and rN409 ELISAs. This may be because the rN160 recombinant protein has the smallest molecular weight among the three recombinant proteins, which results in a minimal nonspecific reaction with chicken sera. The rN160 ELISA also had high specificity and sensitivity, as well as a strong significant positive correlation with a commercial ELISA kit based on the whole virus. The discordant positive results might be ascribed to some differences in the viral antigen preparations adsorbed onto ELISA microplates and to the predominant antigen in rN160 ELISA (N protein), which is more antigenic, immunogenic, and cross-reactive, and reacts with the antibody produced earlier during a humoral immune response.
An ideal antigen for a serological test requires a readily available preparation of pure antigen (Ndifuna et al., 1998). To establish a cost-effective and specific method for IBV diagnosis, the robust prokaryotic host E. coli was commonly used for protein production. Because E. coli is a common commensal microorganism of all animals, it is reasonable to suspect that most field serum specimens will contain antibodies against E. coli; hence, the purity of the coating antigen expressed in such a host might affect the diagnosis accuracy.
Contamination of the coating antigen with E. coli proteins has been proposed to lead to false positives when serum-diagnosing animal diseases (Wang and Khan, 2000). GST affinity chromatography or preabsorption of the tested serum specimens with E. coli crude extract has been suggested as ways of eliminating the false positive problem. In this study, the rN160 protein showed solubility expression in the E. coli system, and one GST affinity chromatography run was sufficient to purify the recombinant proteins to high purity (∼90–95%). Hence, the recombinant nucleocapsid protein rN160 in this study can be expressed and purified in an economical and reproducible manner.
The rN160 ELISA could serve as an ideal serological method for detecting IBV-specific antibodies in chicken sera because of its high cross-reactivity with sera against different IBV genotypes, likely due to the high amino acid sequence similarity of the 1–160 region in the N gene between the CK/CH/LJL/04I and other IBV strains. In addition, the established rN160 ELISA has a high level of agreement for both clinical serum samples and sera from vaccinated chickens under experimental conditions compared with the commercial ELISA kit with whole virus as the coating antigen. To the best of our knowledge, this is the first time that a rapid, simple, and sensitive ELISA has been developed using a recombinant protein from the N protein as the coating antigen to detect group-specific IBV antibodies for epidemiological investigation and antibody-level monitoring.
Footnotes
Authors' Contributions
S.L.: Conceptualization (lead) and writing-review and editing (lead); Y.Z.: Conceptualization (supporting); writing-original draft (lead); and writing-review and editing (lead); Z.H.: Methodology (lead); conceptualization (supporting); and writing -review and editing (supporting); and H.L.: Methodology (supporting) and writing -review and editing (supporting).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Natural Science Foundation of Heilongjiang Province of China Grant (Innovative Research Team Program), China Agriculture Research System of MOF and MARA (CARS-40), and the National Natural Science Foundation of China (32102650).