
Abstract
Chronic hepatitis C virus (HCV) infection poses a major health risk worldwide, with patients susceptible to liver cirrhosis and hepatocellular carcinoma. This study focuses on the development of effective therapeutic strategies for HCV infection through the investigation of immunogenic properties of a DNA construct based on the NS3/4A gene of HCV genotype (g)3a. Gene expression of the mutagenized (mut) NS3/4A target genes was assessed through reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and Western blot analysis. Additionally, bioinformatics tools were employed to evaluate the impact of the mut-NS3/4A-based DNA vaccine. Analysis revealed increased mut-NS3/4A mRNA levels and target protein abundance compared with the native sequence. Elevated mut-NS3/NS4A levels could result from increased RNA stability and proper protein folding. Physicochemical analyses of the protein demonstrated favorable attributes such as thermostability and solubility. Three-dimensional mut-NS3/4A protein modeling confirmed its high stability and agreement with known protein structures. Additionally, potential immunogenic regions of both T and B cell epitopes were discovered based on peptide binding to major histocompatibility complex molecules of Asian origin. Importantly, these epitopes exhibited nonallergenic and nontoxic characteristics. These findings highlight the potential of the NS3/4A-based DNA construct as a promising candidate for an HCVg3a vaccine tailored for the Asian population, providing valuable insights for future immunotherapeutic approaches.
Introduction
Chronic hepatitis C virus (HCV) infection is a global health threat, characterized by lifelong viral persistence and mild clinical symptoms during the acute phase. Most infected individuals progress to chronic HCV infection, which can lead to cirrhosis and liver cancer within two decades if left untreated. Globally, hepatocellular carcinoma accounts for over 17 million cases and 3 million deaths annually (Perz et al., 2006; Posuwan et al., 2019). The global prevalence of HCV infection is estimated at 2.5%, with significant regional variations (World Health Organization, 2024). The Eastern Mediterranean and European regions have the highest number of infected individuals (12 million), followed by the Western Pacific (10 million) and Southeast Asia (similar to Western Pacific). In Thailand alone, an estimated 760,000–790,000 people have tested positive for HCV, with half progressing to chronic infection (World Health Organization, 2024). While HCV infections are declining in developed countries, the mortality rate from HCV-related causes is expected to rise over the next 20 years (Petruzziello et al., 2016). Vaccines that could effectively curb the uncontrolled spread of HCV infection are urgently needed.
Despite significant efforts, achieving complete HCV elimination remains a challenge. Several factors hinder vaccine development, including substantial diversity between HCV genotypes (Smith et al., 2014; Tarr et al., 2015) and the virus’ immune evasion mechanisms (Pantua et al., 2013; Pierce et al., 2016). HCV’s rapid mutational changes pose a challenge for vaccines targeting specific strains. Ideally, vaccines against viruses with multiple strains or variants target conserved epitopes-unchanging regions across various pathogen strains. Utilizing conserved epitopes is a key factor in promoting broad-spectrum immune responses against HCV.
DNA vaccines have shown promising results in animal models for various infectious diseases, including HIV, tuberculosis, leishmaniasis, and toxoplasmosis (Ghaffarifar, 2015; Ghaffarifar et al., 2013; Lee et al., 2004; Plotkin, 2014; Rafati et al., 2001; Taracha et al., 2003). These vaccines encode genetic instructions that mimic the virus and activate the immune system through both major histocompatibility complex (MHC) class I and II pathways. While offering advantages such as eliciting both humoral and cellular immune responses, DNA vaccines can still face limitations in human immunogenicity. To address this limitation, DNA mutagenesis is being investigated as a strategy to enhance antigen immunogenicity. Furthermore, DNA vaccines can also effectively target multiple antigenic targets and strain variants by incorporating different viral proteins or epitopes from various strains into a single vaccine (Encke et al., 2007; Sabet et al., 2014). HCV’s nonstructural protein (NS)3 is a potential vaccination target due to its limited genetic diversity and large size. Mutations in the NS3 protein can enhance its immunogenic properties, leading to a stronger immune response against NS3 (Cheng, 2014; Feng, 2012; Liu, 2018; Zhang, 2016). The resolution of infection has been shown to correlate with induced humoral and specific T cell responses (Diepolder et al., 1997). Enhancing NS3′s immunogenicity by linking it with the NS4A cofactor as a complex leads to increased expression and enhanced stability of NS3 (Frelin et al., 2003; Tanji et al., 1995; Wölk et al., 2000). The NS3/4A-based vaccines were found to be safe with transient effects on viral load and capable of eliciting immune responses both humoral and cellular immune responses in animals, including humans (Ahlén, et al., 2014; Ahlén, et al., 2007; Frelin et al., 2004).
The ability to stimulate immune responses varies among different epitopes present in a pathogen (Babai et al., 1999; Nemchinov et al., 2000). In some cases, vaccines containing only B cell epitopes may not elicit strong immune responses due to a lack of T cell activation (Fujita and Taguchi, 2011). An ideal multisubunit vaccine would contain highly immunogenic T cell epitopes conserved across all HCV genotypes, along with B cell epitopes, to trigger a comprehensive immune response. Recent advancements in bioinformatics offer powerful tools for selecting and predicting potential epitopes that can activate strong cell-mediated and humoral immune responses while also assessing allergenicity and toxicity.
This study aims to utilize bioinformatics to evaluate the impact of mutagenesis on the NS3/4A-based DNA vaccine from HCV genotype 3a, the most prevalent genotype identified in our previous survey. Additionally, bioinformatics will be employed to select T cell and B cell epitopes and analyze their antigenicity, allergenicity, and toxicity. The information obtained will guide the development of multiepitope vaccines against HCV.
Materials and Methods
Construction of recombinant plasmids containing NS3/NS4A
HCV RNA from HCV g3a was extracted from positive sera samples using the QIAamp Viral RNA Mini Kit, following the manufacturer’s instructions (Qiagen, Venlo, Netherlands). The first strand cDNA sequence of HCV g3a was synthesized using Oligo(dT)20 through the RT-PCR SuperScrip® III First-Strand Synthesis System (Invitrogen, San Diego, California, USA), following the manufacturer’s protocol. Gene cloning involved the inclusion of forward and reverse primers with BamHI and XbaI restriction sites at the 5′ and 3′ ends, respectively.
Two separate PCR reactions were performed in a total volume of 25 µL, with cDNA templates, 1× GC buffer, 0.2 mM dNTPs mix, 1 U of Phusion DNA Polymerase (Thermo Scientific, Waltham, Massachusetts, USA), and 0.5 mM forward and reverse primers designed to bind specifically to NS3 gene (F1 containing the Kozak sequence and ATG and R1) and NS4A gene (F2 and R2 containing the stop codon) (Table 1). The PCR conditions included an initial denaturation at 98°C for 30 sec, followed by 35 cycles of denaturation at 98°C for 10 sec, annealing at 52.6°C and 63.6°C, respectively, for 30 sec, and extension at 72°C for 2 min (40 cycles/kb). The final extension was performed at 72°C, 10 min. The 15–40 ng of cDNA products of the two reactions were subjected to the second round of PCR using primer F1 and R2 as above condition except for annealing at 72.3°C. The resulting cDNA fragment of 2.1 kb containing NS3 and NS4A genes was inserted into a BamHI- and XbaI-digested pcDNA3.1+ vector (Invitrogen, San Diego, California, USA).
Primers Used in Gene Cloning and Expression
BamHI: GGATCC XbaI: TCTAGA.
Start codon: ATG stop codon: CTA.
Kozak sequence: GCCACC.
The synthetic mutagenized (mut) NS3/4A genes of HCV g3a were synthesized (Gene Universal, Newark, Delaware, USA). Of the 1908 nucleotides, only 3 were altered (0.15%). Specifically, at position 868, a T was changed to a C, resulting in a codon change from TTG to CTG. Importantly, this change does not affect the amino acid sequence, as both codons encode leucine (L). At position 875, a T was changed to a C (TTC to TCC). This alteration does change the amino acid from phenylalanine (F) to serine (S). Similarly, at position 955, a T changed to a C (TCC to CCC), changing the amino acid from serine (S) to proline (P). The overall impact on the protein sequence was only 2 out of the total of 637 amino acids in the NS3/4A protein. The mutagenized NS3/4A DNA sequence was inserted into a BamHI- and XbaI-digested pcDNA3.1+ vector (Invitrogen, San Diego, California, USA). These plasmid constructs were grown in Escherichia coli, JM109 strain, and screened on LA containing 100 µg/mL ampicillin. Recombinant colonies were verified using BamHI-XbaI double digestion and PCR. DNA sequencing was performed to confirm the correct sequence and reading frame (Integrated DNA Technologies, Iowa, USA). The concentration of the selected plasmids was determined by a nanodrop (ND-2800-ODJ Nano DOT Nucleic Acid Analyzer, Hercuvan Lab system, Malaysia) before protein expression analyses.
In vitro transcription and translation
The gene expression capacity of the constructed plasmids was evaluated through in vitro translation using the prokaryotic T7 coupled reticulocyte lysate system (TNT) (Promega, Madison, Wisconsin, USA). The reactions were carried out in a total volume of 50 µL, comprising 1 μg of each construct and TNT® T7 master mix, methionine, and [35S] methionine, following the manufacturer’s instructions. The reactions were incubated at 30°C for 60–90 min. The pcDNA3.1+ empty vector was utilized as a negative control in these experiments.
HepG2 cell transfection
Prior to transfection, 1.3–2.0 × 105 HepG2 cells (ATCC HB-8065) were plated in a 24-well plate with 1 mL of DMEM low glucose serum-free medium, containing 1× penicillin-streptomycin, 0.55 μg/mL fungizone, and 0.01 M herpes, pH 6.8. The cells were incubated at 37°C and 5% CO2. For transfection, 100 μL of serum-free medium was mixed with the plasmid constructs containing HCV g3a NS3/4A or mut-NS3/4A DNA sequences. Then, 2 μL of Turbofect transfection reagent (Thermo Scientific, Waltham, Massachusetts, USA) was added and vortexed thoroughly. In parallel, the pcDNA3.1+ empty vector was used as the negative control. The mixtures were incubated at room temperature for 15–20 min and transferred to cleaned HepG2 cells in the wells. The culture plates were incubated at 37°C and 5% CO2 for 24, 48, and 72 h. At the indicated time points, cells were detached with 200 μL of trypsin and harvested by centrifugation at 1500 rpm for 5 min. The cell pellets were then extracted for proteins by adding 50 μL RIPA buffer (Thermo Scientific, Waltham, Massachusetts, USA). Before determining the protein concentration using a PierceTM BCA protein assay kit (Thermo Scientific, Waltham, Massachusetts, USA), an equal volume of 10 μL/mL Halt protease inhibitor cocktail (Thermo Scientific, Waltham, Massachusetts, USA) was added.
NS3/4A gene expression analysis by two-step RT-qPCR
The relative mRNA expression level of the NS3/4A gene was determined using a two-step RT-qPCR assay. Transfected HepG2 cells of 50–100 mg were extracted for the total RNA with Trizol reagents according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The first strand of cDNA of the NS3/4A gene was synthesized using SuperScript® III first-strand synthesis system for RT-PCR (Invitrogen, San Diego, California, USA). The reaction comprised 5 µM Oligo (dT) primers, 1 mM dNTPs mix, and 1 pg-5 µg total RNA in the total volume of 10 µL. The mixtures were incubated at 65°C for 5 min to denature the secondary structure of RNA. After preserving the denatured structure on ice for 2 min, the reactions were performed by adding 1× RT buffer, 5 mM MgCl2, 2 U RNaseOut, and 10 U SuperScript® III in the total volume of 20 µL. The first strand cDNA was synthesized at 50°C for 50 min. The reactions were stopped by incubation at 85°C for 5 min, and RNA templates were subsequently eliminated with 0.1 U RNaseH, incubated at 37°C for 20 min.
The first cDNA strand was used as a template for PCR. According to the manufacturer’s instructions, the reaction comprised 1× GC buffer, 0.2 mM dNTP mix, 0.5 mM forward and reverse primers specific to HCVg3a NS3/4A, and 0.01 U Phusion DNA polymerase [Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific, Waltham, Massachusetts, USA)]. The parallel experiments using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were included as the external control. The reactions were performed at 60°C annealing temperatures for HCV NS3/4A and GAPDH. The amplified products were visualized on 1% agarose gel electrophoresis. The cDNA amplified products were used as templates for qPCR reaction (Luna® universal qPCR master mix) (New England Biolabs, Ipswich, Massachusetts, USA). The reactions comprised 1× qPCR master mix, 0.25 µM of the pair of forward and reverse primers, and 30 ng of cDNA template in a total volume of 20 µL. The reactions were the initial denaturation at 95°C for 10 min, denaturation at 95°C for 1 min, and annealing/extension at 60°C sec for 45 cycles. The relative expression ratio (R) of NS3/4A mRNA compared with GAPDH was calculated using the ΔΔCt method (Formula 1).
Dot blot and the Western blot
Dot blot analysis was employed to screen the optimal protein expression of HCV NS3/4A genes through time-course experiments. Twenty nanograms of extracted protein from transfected HepG2 cells were spotted on a nitrocellulose membrane and baked in an oven at 80°C overnight. The membrane was blocked with a blocking solution for 4 h, and an NS3 gene-specific monoclonal antibody (NS3-sc-69938, Santa Cruz Biotechnology) was added at a dilution of 1:200 in PBS buffer. After incubation at room temperature with slight shaking for an hour, the optimized HRP reagent working dilution (Piece Fast Western Blot Kit, Super Signal West Pico Substrate) (Thermo Scientific, Waltham, Massachusetts, USA) was added and incubated at room temperature for 15 min. After washing, KPL 1-Component TMB Membrane Peroxidase Substrate (SeraCare Life Sciences, Milford, Massachusetts, USA) was added to the membrane and incubated at room temperature for 10 min or until color spots were observed. The reactions were stopped by rinsing with clean water for 20–30 sec. The 50 μg of expressed proteins were further verified by Western blot analysis. The expressed proteins were separated on a 12% gel compared with the Precision Plus Protein Dual Color Standard protein marker (Bio-Rad, Hercules, California, USA). The protein bands on a replica gel were transferred onto a nitrocellulose membrane in transfer buffer (192 mM glycine, 25 mM Tris 10% (v/v) methanol) at 25 V overnight. The Western blot protocol was similar to that described above, but a 1:1000 anti-β actin antibody (Abcam, Waltham, Massachusetts, USA) was added as an endogenous control. SDS-PAGE and the Western blot analysis analyzed the resulting protein products of approximately 68 kDa. The ratio of absolute intensities between the target protein and beta-actin, referred to as “band intensities relative to beta-actin,” was utilized to assess protein expression differences between the native sequence and mut-NS3/4A.
Analysis of the mutagenized gene
Codon usage bias can impact various genomic characteristics, including GC content (Fuglsang, 2003; Spencer et al., 2012; Ward et al., 2011) and the efficiency of translation (Hiraoka et al., 2009; Welch et al., 2009).
The Bioinformatics Center of Biologics International Corp (https://www.biologicscorp.com/) was used to analyze codon usage bias in the NS3/4A target gene (Supplementary Table S1). Codon adaptation index (CAI) and GC content were evaluated to assess potential expression levels. An RNA sequence with high CAI is expected to have higher rates of translation (Leppek et al., 2022; Reinhard et al., 2020). For optimal gene expression level prediction, the GC content should ideally be in the range of 30–70%, while a CAI score between 0.8 and 1.0 (Sharp and Li, 1987).
Prediction of RNA secondary structure
RNA secondary structures impact translation efficiency and protein production (Ahmad et al., 2016). The Vienna RNA Web Services (RNAfold web server) (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) was used to predict the most stable secondary structure for the mRNA sequence, based on minimum free energy (MFE). A lower MFE indicates a more stable structure.
Physicochemical parameter prediction of proteins
The physicochemical properties of a vaccine candidate influence its stability, solubility, and immunogenicity. The Expasy ProtParam tool (https://web.expasy.org/protparam/) was used to calculate properties like molecular weight, theoretical isoelectric point (pI), and amino acid composition. A high aliphatic index indicates greater thermostability, while a low instability index (II) suggests a stable protein (Khalid and Ashfaq, 2020). The GRAVY (Grand Average of Hydropathy) score provides insights into hydrophobicity, which can affect protein solubility and interaction with immune cells. Protein solubility was additionally assessed using the Protein-Sol solubility prediction server (https://protein-sol.manchester.ac.uk/).
Three-dimensional structure prediction of optimized proteins
The three-dimensional (3D) structure of a protein is crucial for understanding its function and potential interactions with the immune system. The Expasy SWISS-MODEL (https://swissmodel.expasy.org/) was used to generate a homology model of the optimized protein. The optimal templates were selected based on the Global Model Quality Estimate (GMQE) score, which combines properties from the target-template alignment and the template structure. An overall model quality ranges between 0 and 1, where greater values suggest better quality. Model validation was carried out in MolProbity, which is a score that combines the clash score, rotamer, and Ramachandran evaluations of predicted protein structure into a single score, all normalized to the X-ray resolution scale. A value −4 < x < 2 is considered appropriate for normal structures. Clash score is the number of serious steric overlaps (>0.4 Å) per 1000 atoms of the predicted protein structure. The lower number is better, while a higher percentile is better.
Subsequently, the models were refined using the GalaxyRefine2 server (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE2). The quality and accuracy of 3D protein structure were evaluated using four program web servers including ProSA analysis, ERRAT, Vadar, and MolProbity. The ProSA-web tool (https://prosa.services.came.sbg.ac.at/prosa.php) was utilized to assess potential errors in 3D models using the Z-score index. ERRAT (https://servicesn.mbi.ucla.edu/ERRAT/) was used to analyze the statistics of nonbonded interactions between different atom types in the 3D model. An average overall quality factor of around 91% is considered acceptable for structures with low resolutions. The Ramachandran plot in Vadar Version 1.8 online software (http://vadar.wishartlab.com/) was used to visualize and evaluate the Ramachandran’s plot of the predicted model. Additionally, MolProbity (http://molprobity.biochem.duke.edu/) was also used to analyze the geometry without all-atom contacts of the model. All programs were run as default parameter.
T cell epitope prediction
Cytotoxic T lymphocytes are essential for defense against intracellular pathogens. The most selective stage in the presentation of the antigenic peptide to the T cell receptor is the binding of the MHC molecule. The lengths of the epitopes were set to 9.0. The prediction of MHC class I-restricted T cell epitopes included MHC-I binding prediction, transport efficiency, and proteasomal C-terminal (CT) cleavage prediction. MHC class I-restricted T cell epitope prediction focused on 10 MHC I supertypes prevalent in Asian populations (Supplementary Table S2). The Immune Epitope Database (IEDB) analysis tool (IEDB-AR) v.2.22 (http://tools.iedb.org/main/), NetMHCpan-4.1 (https://services.healthtech.dtu.dk/services/NetMHCpan-4.1/), and Rankpep (http://imed.med.ucm.es/Tools/rankpep.html) were used to identify epitopes with high binding affinity to MHC molecules. Strong binders are defined as having a percentage rank <0.05. The top high-scoring epitopes with the least percentile rank in binding affinity were selected. Furthermore, Epijen v 1.0 (http://www.ddg-pharmfac.net/epijen/EpiJen/EpiJen.htm) was used to assess intrinsic potential of T cell epitopes based on cleavage sites and TAP binding affinity.
Class II-restricted T helper cells mediate the growth and differentiation of both T effector cells and antibody-producing B lymphocytes. MHC-II binding predictions were performed using the IEDB analysis resource MHC-II binding tool (http://tools.iedb.org/mhcii/). This type of epitope prediction was performed using DRB1, MHC class II supertype in Asian countries known for its role in protecting against HCV (Supplementary Table S3). The default setting was to choose 15 mer lengths for the epitopes, and the resulting epitopes were categorized based on the percentile value. Epitopes with the lowest consensus scores, indicating strong binding affinity, were selected. These epitopes were also assessed for homology with human proteins using BLAST to minimize the risk of autoimmunity.
B cell epitope prediction
Antibodies recognize B cell epitopes, which can be composed of either linear peptide sequences or conformational determinants. Conformational determinants are only present in the 3D form of the antigen. The 20-mer linear B cell epitopes were predicted using different servers: IEDB servers (http://tools.iedb.org/main/bcell/) and the ABC Pred server (https://webs.iiitd.edu.in/raghava/abcpred/), which utilizes an artificial neural network (machine-based technique). Furthermore, the Ellipro server (http://tools.iedb.org/ellipro/) was used to predict conformational epitopes from the protein’s 3D structures. Ellipro does not require training datasets and works based on the geometrical properties of protein structure. The default settings were used, with a minimum score of 0.5 and a maximum distance of 6 Å. The predicted epitopes were evaluated based on the residual protrusion index, protein shape, and neighbor residue clustering.
Antigenicity
Good candidate vaccines must possess antigenicity and stability to combat infectious pathogens effectively. VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was used to predict the capability of the epitopes to elicit an immune response within hosts. This tool uses a novel alignment-free method based on physicochemical properties. The prediction threshold influences the accuracy, sensitivity, and specificity of a prediction. The prediction threshold was set at 0.4 to optimize the server’s prediction accuracy. All programs were run as default parameter.
Allergenicity and toxicity determination of T cell epitopes
In addition to efficacy in eliciting an immune response, T and B cell epitopes must be safe for hosts. The allergenicity determination of epitopes, assessing the potential to produce allergenic reactions or hypersensitivity, was carried out using the AllerTOP v. 2.0 online software (https://www.ddg-pharmfac.net/AllerTOP/). The ToxinPred2 online server (https://webs.iiitd.edu.in/raghava/toxinpred2/batch.html) was used to determine toxicity and assess cross-reactivity or tolerance of the epitopes within the hosts. All programs were run as default parameter.
Results
Expression of the NS3/4A-based DNA construct in HepG2 cell lines
Expression of the NS3/4A-based DNA construct in HepG2 cell lines was achieved by cloning the NS3/4A and mut-NS3/4A target genes into the pcDNA3.1+ vector. These recombinant plasmids, along with the empty vector, were successfully transfected into HepG2 cell lines. Gene expression was evaluated using a dot blot assay with an anti-NS3 monoclonal antibody. Dark spots were observed and recorded on the third day posttransfection. Cell cultures on the indicated day were subjected to analysis for mRNA and protein expression using two-step RT-qPCR and Western blot analysis, respectively. The results revealed a 2.5-fold change in mut-NS3/4A mRNA levels (Fig. 1A), while the target protein levels were 7.5-fold higher relative to the native sequence (Fig. 1B and Supplementary Fig. S1).

DNA mutagenesis enhanced NS3/4A gene expression. HepG2 cells were transfected with pcDNA3.1+ containing either the mutagenized NS3/4A (mut-NS3/4A) gene or the wild-type NS3/4A gene. Cells were harvested 3 days posttransfection. pcDNA3.1+ alone (empty vector) served as a negative control.
Analysis of the nucleotide sequences
Nucleotide sequence analysis indicated no significant difference in the G+C content between mut-NS3/4A and NS3/4A (54% and 53%, respectively). Both sequences fell within the optimal G+C content range of 30–70%. However, the CAI value of 0.71 is slightly low, ideally falling within the range of 0.8–1.0. This result indicates that our mut-NS3/4A sequence is reasonably well-adapted but may not be optimal for expression in a human cell as a host organism. Improving the CAI by substituting less-favored codons with those that are more frequently used in the host organism could enhance protein production. However, CAI is just one factor influencing gene expression. Other elements, such as mRNA secondary structure (Mauger et al., 2019), GC content, and regulatory elements, also play significant roles in the overall expression of the gene.
Prediction of RNA secondary structure
The mRNA secondary structures were predicted using the RNA Fold web server. The optimized mRNA had a stable structure with a MFE of −754.1. These results aligned with the assembly diversity (395.3) and mRNA equilibrium base-pairing probability. The predicted secondary structure is shown in Supplementary Fig. S2. The most stable mRNA has mostly double-stranded regions with a few loop regions, contributing to its stability.
Physicochemical properties of the mutagenized NS3/4A construct
Various physicochemical properties of the mut-NS3/4A protein construct were computed using the ProtParam server. The protein had a molecular weight of 68067.7 g/mol and a slightly acidic pI of 6.03. The estimated half-life of the recombinant protein was 30 h, suggesting good stability in mammalian reticulocytes (in vitro). The aliphatic index and the instability index were 83.28 and 39.74, respectively, indicating that the vaccine was moderately thermostable. The GRAVY score of 0.004 indicated a slight hydrophobicity. A Protein-Sol solubility value of 0.259 indicates that the predicted protein had low solubility, which could impact its formulation, stability, and downstream processing.
Modeling of 3D vaccine structure
A 3D model of the mut-NS3/4A protein was generated using SWISS-MODEL, a program adapt at creating protein homology models at various complexity levels. The model was further refined through the GalaxyRefine server (Fig. 2A). Analysis of the model indicated high stability and confidence, supported by a GMQE score of 0.82 and a QMEANDisCo Global score of 0.83 ± 0.05 (both scores measure model quality). Additionally, the MolProbity score (1.26) and Clash score (0.35) suggested good geometry and minimal steric clashes within the protein structure. These scores all improved after refinement, indicating an enhancement in the overall quality of the NS3/4A protein model.

The predicted three-dimensional (3D) structure of the mut-NS3/4A protein. Panel
Ramachandran’s plot analysis, which evaluates the conformation of amino acid residues in protein structures, revealed that the majority of residues (99.13%) occupied favorable or allowed regions, with only a small fraction (0.17%) falling in the disallowed region (Fig. 2B and Supplementary Table S4). This indicates a high-quality model. Further validation using ProSA analysis yielded a low Z-score (−9.52), suggesting a high degree of similarity between the model and known protein structures in the reference set (Fig. 3A). An ERRAT score of 93.053 further reinforced the strong overall quality and alignment of the model with established protein structures (Fig. 3B). These validation results strongly support the reliability of the developed structure for further analysis.
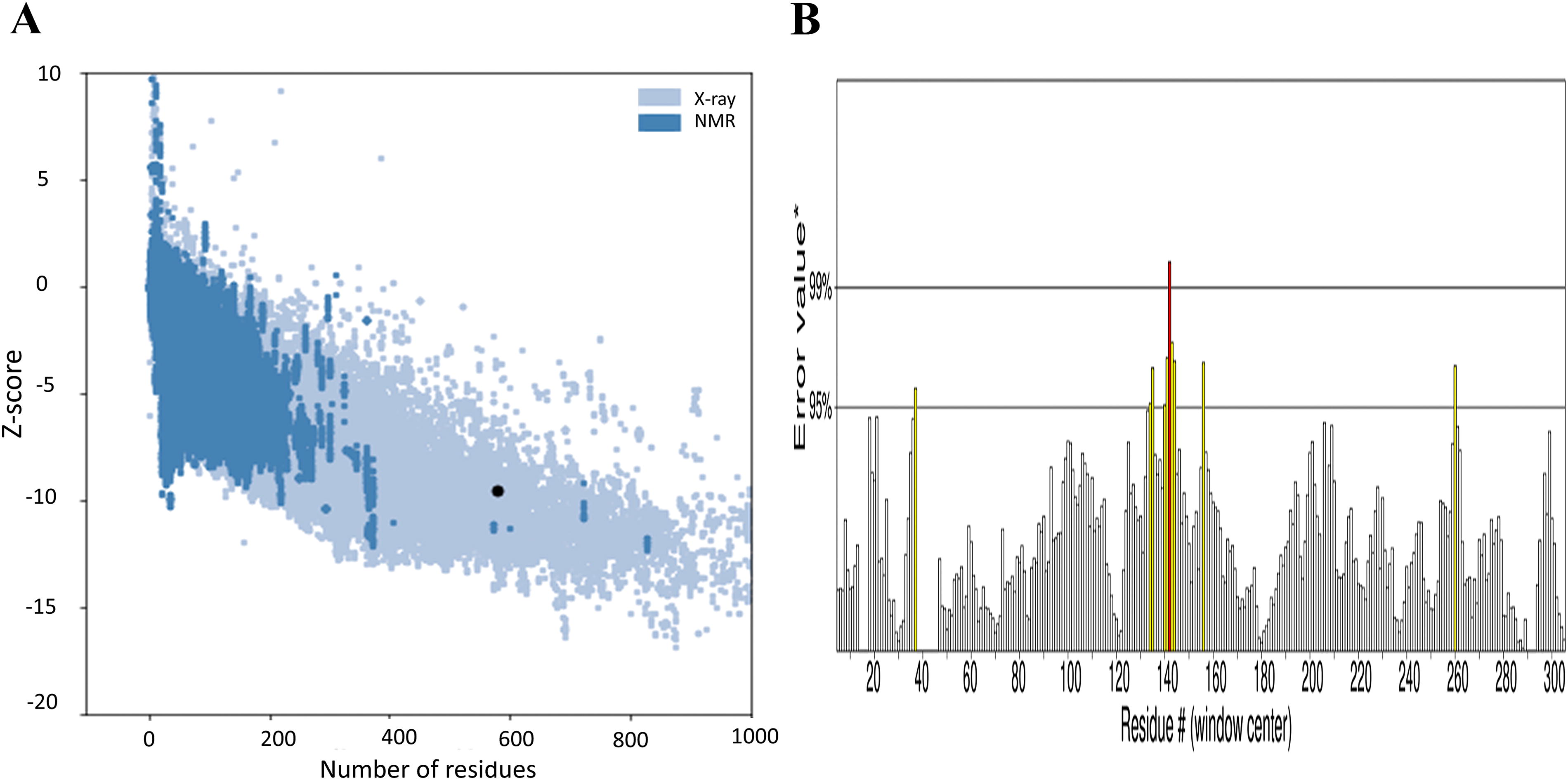
The validation of the predicted 3D structure of the mut-NS3/4A protein. Panel
T cell epitope prediction
T cells play a critical role in eliminating virally infected cells. Their receptors recognize viral peptides presented on MHC supertypes expressed on the surface of infected cells.
We predicted both 9 amino-acid MHC class I and 15 amino-acid MHC class II epitopes. The likelihood of presentation by MHC molecules is influenced by the affinity prediction score, where a lower binding value signifies a higher binding affinity. The analysis also considered potential T cell MHC class I epitopes using scores derived from transporters associated with antigen processing (TAP) transport efficiency and proteasomal CT cleavage prediction. Selection of the best epitopes was based on specific criteria, including 100% conservation among the proteins, optimal binding affinity, high antigenicity/immunogenicity, and no overlap with human proteins. The epitope prediction was limited to 10 MHC I supertypes. Among the 25 epitopes classified as promiscuous binders (Supplementary Table S5), two epitopes exhibited the highest predicted promiscuity: RRGDSTASL at position 67 and GRAIPIALL at position 300. These epitopes can bind to multiple (up to 7) MHC I alleles, particularly those of Asian origin. For the predicted HCVg3 epitope, the top binder was HLA-A01:01 (supertype B15), with a binding percentage of 12.5%. HLA-A02:01 (supertype A2) and HLA-A*31:01 (supertype A3) demonstrated equally good binding, with 10.71% binding each. Six MHC class II epitopes were predicted, but no promiscuous binders were observed (Table 2).
T Cell-Restricted MHC Class II Epitopes Predicted by the IDEB, VaxiJen, Toxipred, and Allertop v2.0
MHC, major histocompatibility complex.
B cell epitope prediction
Twenty-mer B cell epitopes were predicted using IEDB and ABcepred. The VaxiJen server was used to confirm the results, with B cell epitopes scoring above 0.5 selected for further analysis. These analyses revealed six linear B cell epitopes with antigenic properties (TRGVAKALQFIPVETLNTQARSPSFSDNST, SITVPHSNIEEVALGSEGE, LNAVAFYRGLDVSVIPT, DAVSRSQRRGRTGRGKH, HIDAHFLSQTKQSGENFAYL, and ALIPDREVMYQQNDEMEE), as shown in Table 3. In addition to the linear B cell epitopes, the Ellipro server predicted four conformational B cell epitopes (Supplementary Table S6). A 3D model of these conformational epitopes is presented in Figure 4. (The yellow surface in Fig. 4 indicates the discontinuous B cell epitope residues displayed in Supplementary Table S6.)

The 3D conformational B cell epitope of the mut-NS3/4A protein performed by the Ellipro server. The yellow surface indicates discontinuous B cell epitope from A to D following residues displayed from 1 to 4, respectively, in Supplementary Table S6.
Linear B Cell Epitope Predicted by IEDB and ABcepred
IEDB, Immune Epitope Database.
Selection of epitopes and safety considerations
To ensure a safe and effective vaccine, T cell epitopes (both MHC class I and II) were selected based on two key criteria: Overlap with B cell epitopes: Epitopes that overlapped with the previously identified B cell epitopes were prioritized. Similarity to other HCV genotypes: The epitopes were verified for similarity with a diverse range of HCV genotypes (Supplementary Table S7).
Following this selection process, the safety of the candidate epitopes was evaluated using ToxinPred 2.0 and AllerTOP 2.0 online software. Out of the 21 antigenic epitopes identified previously, only eight T cell MHC class I epitopes (TPPAVPQSY, VPQSYQVGY, LGFGSYMSR, GSEGEIPFY, GRAIPIALL, GLNAVAFYR, YRYVSQGER, and ALAAYCLSV) were confirmed to be nonallergenic and nontoxic. For MHC class II epitopes, while six were predicted to be immunogenic, only three (SPSFSDNSTPPAVPQ, GLNAVAFYRGLDVSV, and SGENFAYLVAYQATV) demonstrated the desired safety profile. Encouragingly, all predicted B cell epitopes were classified as nonallergenic and nontoxic.
Discussion
This article presents a comprehensive analysis demonstrating that the mutagenesis of the HCV g3 NS3/4A DNA sequence effectively enhances gene expression at both the transcriptional and translational levels. Bioinformatics investigations further reveal that DNA mutagenesis significantly impacts mRNA secondary structure, stability, translation, and protein folding.
The amount and structure of a vaccine are key determinants for eliciting effective immune responses. Examination of the GC content of the mut-NS3/4A sequence indicates a high probability of expression in E. coli hosts. Additionally, the predicted secondary structure of mut-NS3/4A mRNA suggests that higher mut-NS3/4A mRNA levels could be due to increased RNA stability (Bazzini et al., 2016; Newman et al., 2016; Presnyak et al., 2015; Zhou et al., 2016). An mRNA molecule with increased stability is desirable, potentially resulting in higher potency and improved clinical efficacy. However, our results, along with previous observations (Zhang et al., 2023), demonstrate that the ideal MFE of an RNA sequence may not always align with its CAI.
At the protein expression level, the enhanced mut-NS3/4A protein levels could result from cotranslational protein folding, which influences the speed and accuracy of translation (Presnyak et al., 2015). Ensuring the accuracy of a protein model is essential for reliable vaccine design. Deviations from the Ramachandran plot analysis can indicate structural anomalies, conformational changes (Yılmaz, 2021), misfolded or lower quality structures (Tan et al., 2022), posttranslational modifications (Seadawy et al., 2022), or errors in the experimental data used (Solanki et al., 2019). To mitigate the potential impact of these deviations, careful validation of the experimental data used in the analysis is crucial to ensure accurate determination of the protein’s structure. In this study, the predicted protein was found to have a relatively high overall quality and likely resembles known structures obtained from experiments.
While the DNA mutagenesis and in silico analysis suggest promising aspects of the mut-NS3/4A construct, the analysis of its physicochemical properties reveals limitations. The mut-NS3/4A protein construct is predicted to be slightly acidic, hydrophobic, moderately heat-resistant, and not very soluble. These properties could impact formulation, stability, and downstream processing. Therefore, the construct, based solely on its physicochemical properties, may not be an ideal candidate for an antiviral vaccine.
Vaccines designed to target specific epitopes may offer greater efficacy. The selection criteria for final epitopes in vaccine development include a high antigenicity score, nonallergenicity, nontoxicity, and strong binding affinity. An optimal vaccine should contain all viral antigen components to elicit both humoral and cell-mediated immunity, providing protection against primary and secondary infections. In this study, one T cell epitope (MHC I and II) overlapping with a B cell epitope was chosen. MHC class I epitopes elicit cytotoxicity, while MHC class II epitopes trigger a CD4+ helper T cell response, crucial for creating a strong CD8+ T cell memory. B cell epitopes stimulate humoral immunity. Vaccines with a combination of T cell and B cell epitopes can effectively activate both cellular and humoral immune responses, resulting in a long-lasting immunity. The vaccine containing immunogenic consensus sequence epitopes could prevent significant spread of diverse HCV genotypes. Additionally, the epitopes do not induce autoimmunity, cross-reactivity, or tolerance in the host. However, for appropriate elicitation of an immune response, the interaction between the antigenic molecule and immune receptor molecule is necessary. Therefore, further studies using molecular docking analysis are needed to assess epitope-receptor binding affinities.
Conclusions
The mutagenesis of the HCV g3 NS3/4A DNA sequence effectively enhances gene expression at both transcription and translation levels. Bioinformatics provide an efficient and cost-effective approach to vaccine development, allowing for evaluating vaccine effectiveness, side effects, and toxic effects before its actual development. The identified T and B cell epitopes present valuable candidates for safer, more effective, and precise epitope-based vaccines against HCV, specifically tailored for the Asian population. Further experimental and simulation studies will be essential to validate and refine these findings for potential vaccine development.
Footnotes
Acknowledgments
The authors specially thank Witthaya Poomipak, and Prof. Dr. Sunchai Payungporn, Faculty of Medicine, Chulalongkorn University, for valuable advice on figure designs and Prof. Dr. Vithoon Vaiyanant, Founder and President of the Bangkok Pathology-Laboratory Co., Ltd (BPL), who permitted to collect deidentified blood samples from his company. The authors also thank Assoc. Prof. Dr. Lars Felin and Prof. Dr. Matti Salberg from Krollinska Institutet for providing the vector for DNA cloning and inspiring us to work on this project.
Authors’ Contributions
P.C.: Investigation (equal), data curation (lead), formal analysis (lead), and writing—original draft article (lead). K.S.: Funding acquisition (equal) and investigation (equal), U.I.: Funding acquisition (equal), conceptualization (lead), supervision (lead), methodology (lead), formal analysis (supporting), writing—review and editing article (lead). All authors have read and approved the final version of the article. All authors have accepted responsibility for the entire content of this article and approved its submission.
Availability of Data and Materials
The datasets used and/or analyzed during this study are available from the corresponding author on reasonable request.
Ethics Approval
This research used anonymized blood serum samples obtained from the Bangkok Pathological Laboratory, a reference laboratory that operates privately. These samples were collected following strict protocols to ensure complete confidentiality for the donors. Researchers had no access to any personal identifying information. The study protocol, including the use of these samples, was reviewed and approved by the Institutional Review Board of Burapha University (BUU-IRB), an ethics committee to ensure responsible research practices.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This work was fully supported by National Research Council of Thailand (NRCT) (grant number 2559A10802094).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.