
Abstract
Viral infections are one of the principal causes of global primary health crises, with increased rate of infection and mortality demonstrated by the newer progeny of viruses. Viral invasion of the host involves utilization of various cellular machinery. Ubiquitination is one of a few central regulatory systems used by viruses for establishment of the infections in the host. Members of the ubiquitination system are involved in carrying out proteasomal degradation or functional modification of proteins in numerous cellular processes. E3 ubiquitin ligases play a major role in this system through recognition and recruitment of protein substrates and catalyzing the transfer of ubiquitin to these substrates. The versatility of ubiquitin ligases frequently makes them useful tools for the viruses, for either utilizing or degrading other cellular machineries, for carrying out their multiplication or inactivating the defensive strategies of the host. Therefore, these ligases are important targets for aiming at major pathways causing viral protein degradation or functional modification of the infection process. In this review, we have discussed the role and mechanism of different types of ubiquitin ligases in the context of infections of mainly human viruses, highlighting the viral proteins directly interacting with the ligases. Knowledge about these direct interactions is central in understanding the ubiquitin-dependent processes. This comprehensive account may also be beneficial for pharmaceutical exploration of E3 ligase-based broad-spectrum antiviral treatment.
Introduction
Viruses are ultra-small, obligate intracellular infectious agents that depend on a suitable host cell for their propagation. Each virus infection is guided by the genetic code of the virus itself, while mostly depending on the complex metabolic and biosynthetic machinery of the host (Sela and Hilleman, 2004; Nagy and Pogany, 2011). Host cell responses to these infections vary widely, such as, limiting the disease, immune responses, or contribution to the infection and death, causing various levels of severity of the resulting diseases (Eisfeld et al., 2024; Yang and Yang, 2021). Advances made in genomics, proteomics, and medical sciences improve our understanding of viral nature, genomic machinery, and host pathogenicity and make us capable of countering the majority of epidemics (Artika et al., 2020; Gorbalenya and Lauber, 2022). However, several viral epidemics over the last decades with unexpected mortality, such as Zika, Ebola, and, most notably, novel severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2), have underlined the significance of research and reappraisal on host–virus interaction (Baker et al., 2022).
The complex interaction between the host cell and virus occurs through two main processes—the viral life cycle and the interplay between the virus and the host defense mechanism. Host innate immunity is the frontline defense against viral infection, which is followed by a more specific adaptive immunity. Completion of life cycle of the virus depends on its abilities to avoid or suppress the host defense mechanism—thus, several components of the host innate immunity pathway become targets for one or the other infecting viruses.
Many of the steps in the life cycle of virus and the defense mechanism of the host are regulated by a reversible post-translational modification, called ubiquitination. Ubiquitin is a 76 amino acid protein that is conserved throughout eukaryotes. Conjugation of ubiquitin to a target protein causes changes in the stability and activity of the target (Gu and Jan Fada, 2020). The entire process of ubiquitination consists of three steps, each catalyzed by a specific class of enzymes, ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3), respectively (Yang et al., 2021). E1 activates ubiquitin in an ATP-dependent fashion, E2 identifies the E1-ubiquitin complex and receives the ubiquitin to form a E2-ubiquitin conjugate. E2 then finds the E3, that is attached with substrates and help transferring the ubiquitin to those substrates (Schulman and Harper, 2009; Yang et al., 2021). The E3 can interact with both the charged E2 and the substrate—and catalyzes the final steps of the substrate ubiquitination. Among all three groups of enzymes, E3 ligases belong to the most diverse group of almost 600 members. They play the principal role of interacting with the substrates directly and thus determining substrate specificity (Metzger et al., 2014).
Regulations by ubiquitination or de-ubiquitination are some of the most versatile of all the life processes. These regulations play important parts in different viral infections and related host defense mechanisms. Involvement of ubiquitin E3 ligases in the host–virus interaction comes in different ways. Viruses frequently subjugate one or more members of ubiquitin system at different stages of their infections, including virus entry, genome replication, and virus exit from the infected cell. On the other hand, some of the ubiquitin ligases are involved in the host defense mechanism against viruses, such as cellular stress-induced interferon (IFN) production, host cell apoptosis, chromatin remodeling, etc. In return, viruses devise mechanisms to counteract some of these mechanisms and carry on with their infections (Gu and Jan Fada, 2020). In addition to innate immunity, the components of host adaptive immunity, such as CD4 or MHC-I, also become targets of viruses in ubiquitin-dependent pathways.
The present review focuses on the multiple roles of E3 ligases in the host–pathogen interaction of different human viruses. Attempts have been made to make an account of direct interactions of these ligases with the viral proteins.
Viral Infection-Induced Host Innate Immune Response
Viral induction of host innate immune response involves the pathogen-specific structural patterns present on the nucleic acids or some glycoproteins of the infecting viruses known as pathogen-associated molecular patterns (PAMPs) (Mahla et al., 2013). These are recognized by the pattern recognition receptors (PRRs) of the host cells during viral infection. The three major classes of PRRs recognizing viral PAMPs are (a) RIG-I-like receptors (RLRs), (b) toll-like receptors (TLRs), and (c) a group of structurally unrelated viral DNA sensors, of which cyclic GMP-AMP synthase (cGAS) is a representative member (Kawai and Akira, 2010; Li and Wu, 2021; Suresh and Mosser, 2013). In response to viral infections PRRs activate several kinases of the inhibitor of nuclear factor kappa-B (κB) kinase (IKK) family. These kinases then phosphorylate and degrade inhibitor of nuclear factor kappa B alpha (IκBα), allowing translocation of the transcription factor NFκB to nucleus. Other transcription factors that are phosphorylated by IKK are IFN regulatory factors 3 and 7 (IRF3/7), which are also translocated to nucleus. PRRs also activate the mitogen-activated protein kinases (MAPK), which in turn activate activator protein-I (AP-I). NFκB, IRF3/7, and AP-I induce the expression of IFNs and other cytokines, starting a cascade of antiviral effects (Davis and Gack, 2015).
Type I IFN (IFN-I) family is a multigene cytokine family, important for host defense during viral infection. As a result of induction of IFNs, various IFN-stimulated genes (ISGs) are either activated or repressed. This can lead to positive or harmful effects on host. In the canonical JAK-STAT pathway, the interaction between secreted IFN and its receptor causes activation of the members of the Janus kinase family, which in turn phosphorylate the STAT family of transcription factors. This activation results into the activation of ISGs (Gu and Jan Fada, 2020; McNab et al., 2015). A direct transcriptional target of IFN-I is the tumor suppressor protein p53. In response to oncogene expressions, p53 is activated with the resultant induction of cell apoptosis. This protein was also found to be up or downregulated in different viral infections (Gu and Jan Fada, 2020; Hu et al., 2021; Rivas et al., 2010). Figure 1 schematically represents these innate immunity pathways as the object of different virus and host ubiquitin ligase interactions. Details of these interactions regulating the main role players of the host innate immunity and the viral subversion of the host immunity are described in the following sections discussing the roles individual classes of ubiquitin ligases.
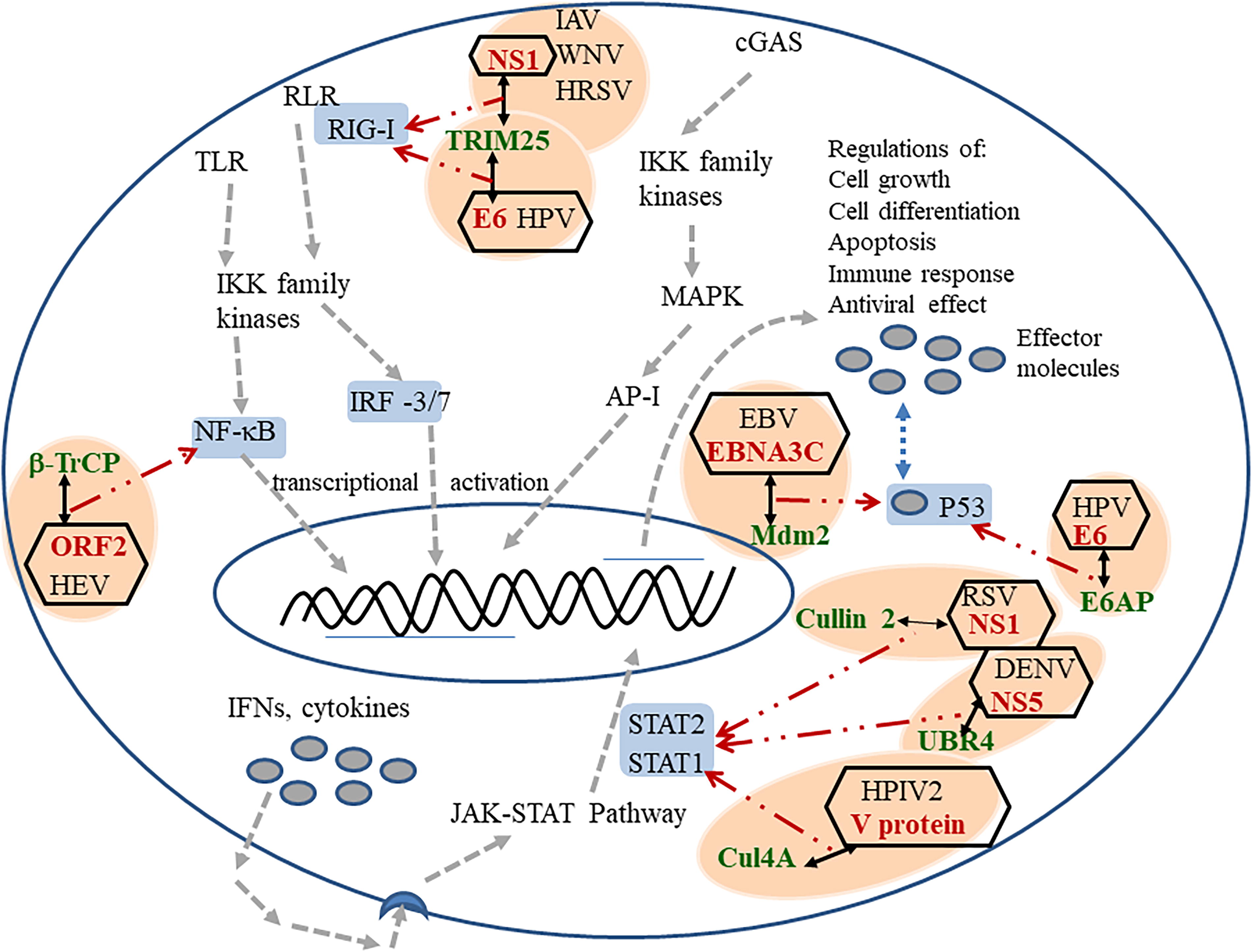
Direct interactions of viral proteins with human E3 ubiquitin ligases, regulating the innate immune response at different points. Host ubiquitin ligases are marked in green color, interacting viral proteins are marked in red color, the important modulators host innate immune response is marked with a gray background, each virus–host protein interaction is marked with an orange oval background, regulation of viral processes are indicated by dashed arrows, positive effect marked by green arrow, negative effects are marked with red arrow.
Types of Ubiquitination and Classes of Ubiquitin Ligases
Ubiquitin has an N-terminal methionine and seven lysine sites that are important for its interaction with substrates; these are K6, K11, K27, K29, K33, K48, and K63. The downstream signaling effects of the ubiquitination depend on the lysine site through which the linkage has occurred. For example, K-48-linked ubiquitination causes proteasmal degradation of the substrate, while K-63 linked ubiquitination causes intracellular signaling in processes such as DNA damage repair, cytokine signaling, etc (Yang et al., 2021).
Based on the number of ubiquitin molecules attached to the substrates, the ubiquitination process can be divided into three categories: (a) monoubiquitination—attachment of a single ubiquitin molecule to the target protein, (b) multiubiquitination—ubiquitination of the target protein at multiple sites, and (c) polyubiquitination—attachment of a chain of ubiquitin to the target protein (Oh et al., 2018; Shoji et al., 2017; Yang et al., 2021). Monoubiquitination is associated with nonproteolytic functional modification, while polyubiquitination generally causes proteolytic degradation.
According to structure and function, E3 ligases are generally divided into four classes: really interesting new gene (RING) finger, homologous to the E6-AP carboxyl terminus (HECT), ring in-between RING (RBR), and U-box type E3 ligases. The sequence similarities among these ligases are low (Young et al., 2021). Mechanistically, the HECT and RBR E3 ligases can transfer ubiquitin directly to the substrate through the formation of a ubiquitin–thioester intermediary on one of the E3 cysteine residues. RING-containing E3 ligases position the ubiquitin-loaded E2 in close proximity to the substrate for E2-to-substrate transfer, which usually occurs on a lysine (K) on that substrate (Metzger et al., 2014; Yang et al., 2021). See Figure 2 for the schematic description of the mechanisms of action of these three types of ligases. Structures of the U-box proteins are similar to RING ligases with slight variation. The following section describes the structural and functional specifications of each of these ligases, as well as the mode of their participation in different viral infections.
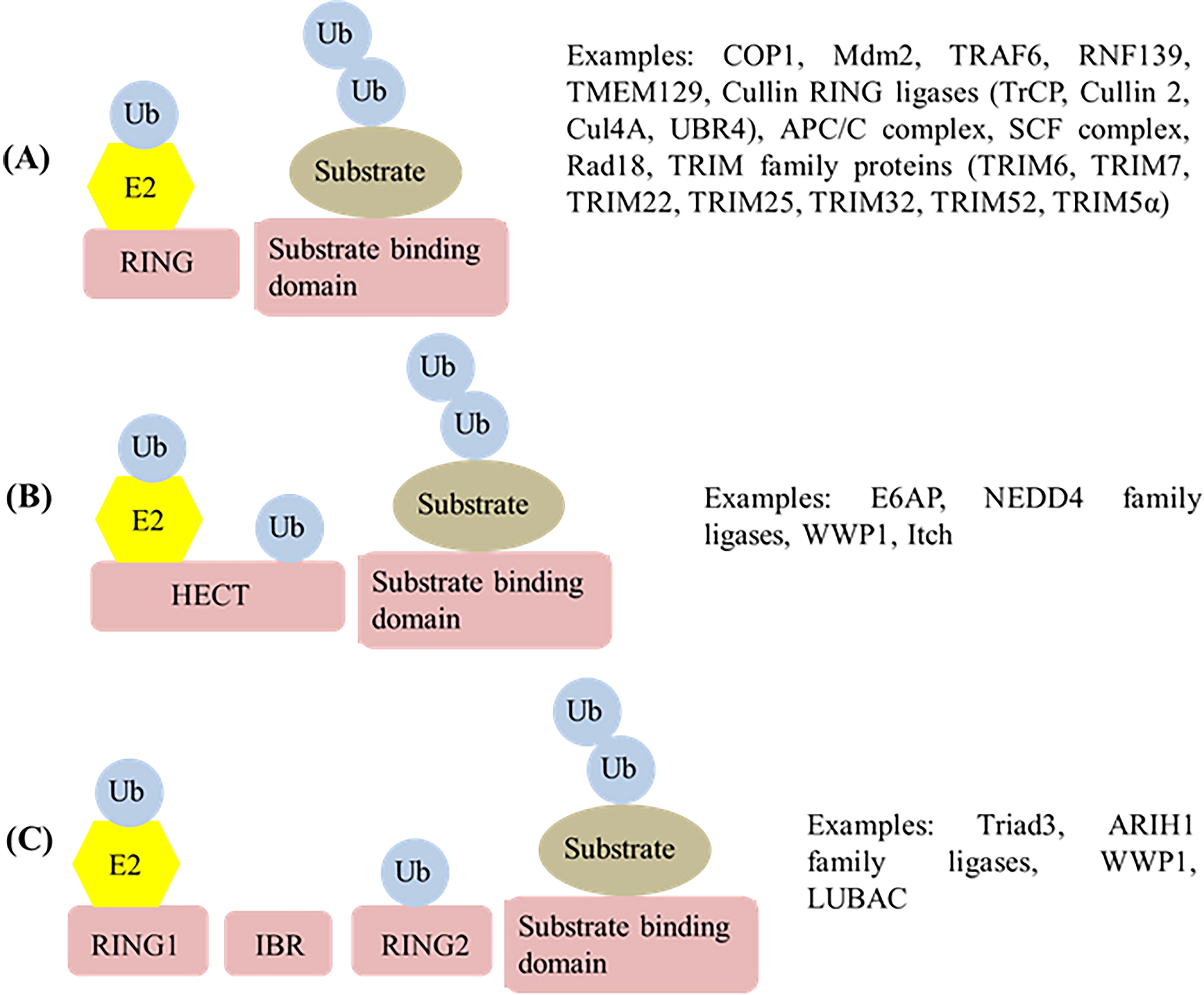
Diagrammatic illustration of different types of ubiquitin ligases and their mechanisms of action, along with examples of the ligases discussed in the present article.
RING E3 Ligases
The RING E3 ligases are the predominant types of E3 ligases expressed in the human body. There are >600 types of RING ligases. They are named after the characteristic RING domain present in their structure. RING domain (also called RING finger or RING motif) is a rigid globular domain containing conserved Cys and His residues engaged in binding two atoms of zinc. This domain is responsible for interaction with E2; it is also the main site for E3 catalytic activity (Deshaies and Joazeiro, 2009; Metzger et al., 2014; Yang et al., 2021). RING E3 ligases belong to two major classes: monomeric and multisubunit E3 ligases. Following is an account of different forms of RING E3 ligases involved in viral infections.
Monomeric RING E3 ligases
In monomeric RING ligases both the substrate binding and catalytic domains are present on the same polypeptide (Metzger et al., 2014; Zheng and Shabek, 2017). Some of the examples of monomeric RING ligases are COP1, Mdm2, and TRAF6.
Mdm2 is known as a key regulator of p53 activities in three main biological processes: aging, cancer suppression, and longevity (Ponnuswamy et al., 2012). As P53 has antiviral effects, many viruses downregulate its activity. Pizzorno et al. (2018) showed that during influenza A virus (IAV) infection the viral nonstructural protein 1 (NS1) modulated P53 activity through destabilization of Mdm2. On the contrary, in Epstein–Barr virus (EBV) infection Mdm2 is stabilized by the viral nuclear antigen 3C (EBNA3C) facilitating the Mdm2 protein-related ubiquitination and downregulation of p53. It was indicated that this regulation of Mdm2–p53 interaction is related to the development of EBV-associated cancers (Saha et al., 2009). Mdm2 was found to regulate the replication of human immunodeficiency virus-I (HIV-I) through its binding with the viral Vif protein and causing its ubiquitination and degradation. Vif is crucial for viral infection as it induces the degradation of one of the host restriction factors A3G (Izumi et al., 2009).
The tax protein of human T-cell leukemia virus type 1 (HTLV-1) was shown to hijack another host RING ubiquitin ligase—the tumor necrosis factor receptor-associated factor 6 (TRAF6). Activation of TRAF6 by Tax causes downstream regulation of several receptors involved in immunoregulation (Walsh, Lee, and Choi, 2015). One of the proteins regulated by TRAF6 is the anti-apoptotic protein MCL-1. The ubiquitination stabilizes and protects MCL-1, thus, is supportive of its role in cell survival and immortalization (Choi and Harhaj, 2014). TRAF6-related ubiquitination was also shown to be a tool used by EBV for upregulation of IRF7, a prime regulator of IFN-α/β responses (Ning et al., 2008). Conversely, degradation of TRAF6 is potentiated by the M1 protein of influenza D virus in order to block IFN signaling. A component of a Cul3 based cullin-RING ligases (CRLs) KEAP1 (Kelch-like ECH associated protein 1), is recruited by M1 for carrying out this ubiquitination (Chen et al., 2022).
RING E3 ligases were found to be involved in the entry of viruses to the host cell and import of viral DNA to the host cell nucleus. E3 ligase c-CBL is generally known to have repressive effect on angiogenesis and regulatory effect on tumor growth (Lyle et al., 2019). It was shown to facilitate entry of Kaposi’s sarcoma-associated herpesvirus (KSHV) to the host cell. This ligase caused monoubiquitination of receptor α3β1, which caused translocation of both the virus and the receptors in lipid rafts causing their macropinocytosis (Chakraborty et al., 2011). Mib1 is another RING-type E3 Ligase whose ubiquitination activity was shown to cause nuclear translocation of adenovirus (AdV) DNA (Bauer et al., 2019). Figure 3 enlists virus and host ubiquitin ligase interactions involved in different stages of viral life cycle, virus entry to host cell, uncoating, replication of viral genome, budding and virus release.

Direct interactions of viral proteins with human E3 ubiquitin ligases, causing regulations of viral entry, genome replication, and viral exit. Host ubiquitin ligases are marked in green color, interacting viral proteins are marked in red color, each viral life process is marked with green oval background, each virus–host protein interaction is marked with a pink oval background, regulation of viral processes is indicated by dashed arrows, positive effect marked by green arrow, and negative effects are marked with red arrow.
RNF139 and TMEM129 are two is an ER resident RING E3 ligase, RNF139 is a negative regulator of cell proliferation through G2/M arrest and cell death. This is hijacked by the US2 protein of human cytomegalovirus (HCMV) for degradation of the MHC-I complex, thus fighting off the adaptive immune response. TMEM129 is recruited by the US11 protein of HCMV for the same purpose (Van den Boomen and Lehner, 2015). Another ER-related RING E3 ligase is synoviolin, which is used by HBV X protein for suppressing glutamate dehydrogenase 1. This suppression causes development of hepatocellular carcinoma (You et al., 2024).
TRIM E3 ligases
Tripartite motif-containing (TRIM) proteins are a subclass of RING ubiquitin ligases, which are characterized by a N-terminal RING domain followed by one or two B-box domains (another zinc binding domain). The B-box domains play important roles in protein ubiquitination (Li et al., 2014). These monosubunit E3 ligases have been linked to the regulation of many cellular processes such as cell growth and development, gene regulation, signal transduction, regulation of cell death, innate immune response, and antiviral activity. These proteins either directly interfere with viral processes or indirectly regulate antiviral pathways mainly through their E3 ubiquitin ligase activities (Rajsbaum et al., 2014). Barr et al. (2008) reported that TRIM22 interfered with the budding of HIV-1 through ubiquitination of the viral Gag particles. RNA viruses, such as encephalomyocarditis virus and influenza virus, were also shown to be the targets of the E3 ligase activity of TRIM22 (Eldin et al., 2009). In an IAV infection, TRIM22 was shown to regulate replication by ubiquitinating the viral nucleoprotein and causing its degradation (Di Pietro et al., 2013). Other TRIM proteins causing inhibition of viral replications are TRIM52 and TRIM32, effective in Japanese encephalitis virus and IAV, respectively. In both of these cases the TRIM proteins cause ubiquitination and subsequent degradation of viral proteins involved in genome replication (Fan et al., 2016; Fu et al., 2015). Huang et al. (2016) reported the antiviral effect of another TRIM protein, TRIM5α, on the proliferation of EBV. TRIM5α was found to promote the ubiquitination of a viral protein Rta (replication and transcription activator), which is involved in the activation of viral lytic gene. The ubiquitination caused a reduction in the transcription activation of Rta.
A number of TRIM proteins were also shown to have proviral effect. In zika virus, the envelope protein E is polyubiquitinated by the host ubiquitin ligase TRIM7. The ubiquitinated E enhances viral attachment to host cell receptor promoting viral entry (Giraldo et al., 2020; Valerdi et al., 2021). The VP35 protein of Ebola virus (EBOV) has multiple roles in viral infection, such as viral genome packaging, acting as essential cofactor of viral polymerase, etc. It was shown that VP35 is ubiquitinated by host ubiquitin ligase TRIM6, which is essential for its interaction with the polymerase, resulting in efficient viral replication, transcription and virus assembly (Bharaj et al., 2017; van Tol et al., 2022). In SARS CoV-2 as well, ubiquitination of the viral NP protein (nucleocapsid protein) by TRIM6 promotes proliferation of the virus (Zhou et al., 2024).
In addition, viral proteins have also been found to target host TRIM proteins in order to carry out infection. Polyubiquitination of RIG-I is a key process in the IFN production and antiviral responses against RNA viruses. In order to prevent this, RNA viruses such as IAV or West Nile virus express their NS1 proteins that interact with TRIM25, one of the ubiquitin ligases causing K-63 linked polyubiquitination of RIG-I (Ban et al., 2018; Gu and Jan Fada, 2020; Rajsbaum et al. 2012). Oncoprotein E6 of human papillomavirus (HPV) was also found to target TRIM25 causing suppression of RIG-I signaling pathway (Chiang et al., 2018).
Multimeric RING E3 ligases
Multisubunit RING E3 ligases are exemplified by CRLs, APC/C, FANC, etc. In these ligases the catalytic and substrate recruiting domains are present in separate protein subunits (Metzger et al., 2014; Zheng and Shabek, 2017). One of the well-studied families of CRLs are SCF (Skp1-Cullin-F-box) E3 ligases. Dysregulation of different proteins of this complex is related to the progression of different types of cancers, such as gastric or prostate. Anaphase-promoting complex or cyclosome (APC/C) plays important roles in cell division and tumorigenesis. Different members of this family have been found to participate in apoptosis, tumor progression, cell cycle regulation, etc (Yang et al., 2021). FANC ligases are involved in DNA repair processes (Metzger et al., 2014).
Many viruses are known to take over CRLs causing oncogenic transformation of the host cells. KSHV causes human malignancies mainly in patients with AIDS. This virus regulates a number of pathways related to cell proliferation and immunity in order to create a cellular environment favorable for persistence of the virus. The latency-associated nuclear antigen (LANA) protein of KSHV binds to a component of a SCF ubiquitin ligase SCFFbw7 and inhibits its function. This prevents ubiquitination of Notch proteins, causing their stabilization, finally facilitating the proliferation of KSHV-infected cells (Ashizawa et al., 2012; Lan et al., 2007; Mahon et al., 2014). Other DNA viruses also cause host cell malignancy by deregulation of CRLs. The oncoprotein E7 of HPV type 16 binds with cullin 2 ubiquitin ligase and activates it. This activation is largely responsible for degradation of the retinoblastoma tumor suppressor protein pRB, causing oncogenic transformation of the host cells (Huh et al., 2007). This oncoprotein shows sequence similarities with the E1A protein of AdV, which binds to and interferes with the function of ubiquitin ligase SCFFbw7, causing oncogenic proliferation of the host cell. Normally SCFFbw7 is a tumor suppressor, which causes degradation of a number of protooncogene products (Isobe et al., 2009). E1A also causes pRB mediated cell transformation. However, this interaction of E1A does not involve ubiquitin ligase activity (Liu and Marmorstein, 2007; McLaughlin-Drubin and Münger, 2009). Oncogenic transformation of host cells by these viruses ensures that host replication machinery remains available for replication of the viral genome (Isobe et al., 2009).
CRLs have also been shown to play essential roles in the infection of DNA viruses, through regulation of viral replication, host immune response, etc. In vaccinia virus (VV) infection, ubiquitination carried out by two components, cullin 3 and Rbx, of a multisubunit CRL system were found to be necessary to initiate viral DNA replication. The substrate for this interaction is not identified so far (Mercer et al., 2012). In EBV, the viral BPLF1(BamH1 fragment left open reading frame-1) protein was shown to stabilize host CRL Rad18, which is important for lytic replication of the virus (Hau and Tsao, 2017; Kumar et al., 2014).
Among retroviruses, HIV-I has been found to hijack host ubiquitin ligases for suppressing host antiviral responses. Human APOBEC3G (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G) is a host cytidine deaminase, which blocks HIV-I replication in certain T cells in the absence of viral protein Vif. In the wild type HIV-I Vif interacts with Cul5 and other host proteins to form a SCF like E3 ligase complex which causes ubiquitination and degradation of APOBEC3G (Iwatani et al., 2009; Yu et al., 2003). RING ligases have been used by viruses also to target the components of adaptive immunity. In HIV-I infection, the viral Vpu protein recruits the host RING E3 ligase SCFβ-TrCP in order to ubiquitinate CD4 leading to its degradation (Margottin et al., 1998; Ramirez et al., 2015)
There are several reports of RNA viruses using host RING E3 ligases for different purposes. In the infection caused by the RNA virus IAV, host Cullin 4-based E3 ligases ubiquitinate subunit PB2 of viral RNA polymerase which contributes to virus production (Karim et al., 2020). Host immune modulations were shown by several RNA viruses, including rotaviruses and reoviruses (Sherry, 2009). In hepatitis E virus infection, the viral ORF2 (open reading frame 2) glycoprotein associates with the βTRCP components of CRL complex SCFβTRCP, causing decreased ubiquitination and degradation of IκBα. Resulting stabilization of IκBα causes suppression of both NFκB signaling and host IFN responses (Surjit et al., 2012). Members of paramyxovirus family, Mumps virus or human parainfluenza virus 2 causes CRL dependent degradation of STAT (signal transducer and activator of transcription) proteins in order to diminish the host IFN responses (Kubota et al., 2005; Ulane and Horvath, 2002). Similarly, in respiratory syncytial virus infection, the viral NS1 protein interact with the host cullin 2 ligases for degradation of the STAT2 protein (Elliott et al., 2007). In case of dengue virus infection, viral NS5 interacts with the host UBR4 E3 ligase causing degradation of STAT2 (Morrison et al., 2013).
The APC/C is a multisubunit RING ubiquitin 3 ligases, which plays an important role in cell cycle regulation. The function of this ligase is inactivated by HCMV. Degradation of the APC components by HCMV protein pUL21a causes transition of the quiescent host cells into S phase and expression of enzymes required for viral DNA replication (Fehr et al., 2012; Wiebusch et al., 2005). The Vpr protein of HIV-I was shown to cause degradation of APC1, a component of the APC/C complex. Exact effect of this degradation on pathogenesis was not clear (Barbosa et al., 2021). AdVs also cause inactivation of APC/C causing apoptosis in the virus infected cells (Heilman et al., 2005).
Viral RING E3 ligases
In addition to host RING E3 ligases, several viruses also encode the RING E3 ligases to facilitate their replication in the host body. For example, the infected cell protein 0 of Herpes simplex virus (HSV)-1 is a RING E3 ligases, which plays a major role during lytic and latent infections (Rodríguez et al., 2020). The K3 and K5 proteins of KSHV are E3 ubiquitin ligases and they cause downregulation of host cell surface MHC-I molecules by facilitating their endocytosis and lysosomal degradation (Boname and Lehner, 2011). Also, KSHV LANA protein acts as a component of a E3 ubiquitin ligase and causes polyubiquitination of P53 (Ashizawa et al., 2012).
HECT E3 Ligases
The homologous to the E6AP carboxyl terminus (HECT) class of E3 ligases derive their name from one of its members E6AP, which forms a complex with the oncogenic protein E6 of HPV. A number of host proteins shows sequence similarities with a 350-amino acid sequence region found near the C-terminal of E6AP (E6-associated protein, also known as UBE3A) (Scheffner et al., 1993; Wang et al., 2020). These HECT proteins are able to form a thioester bond with the C-terminus of ubiquitin and catalyze the formation of polyubiquitin chains. There are 28 such HECT E3 ubiquitin ligases, ranging in size from 80 to 500 kDa, present in human cells and playing prominent roles in disease-relevant processes including viral infection, cancer, neurological disorder, and autoimmune diseases (Wang et al., 2020).
From the perspectives of viral infection and immune response, the E6AP and NEDD4 (neural precursor cell expressed, developmentally downregulated 4) subfamilies are specifically targeted by viral proteins to establish viral replication by overcoming the ubiquitination process and the innate immune response of the host (Davis and Gack, 2015). NEDD4-like ubiquitin ligases have been shown to be involved in the budding process of a number of retroviruses. Yasuda et al. (2002) showed that BUL1, a NEDD4-like ubiquitin ligase interacted with the L domain of Gag protein of Mason–Pfizer monkey virus and facilitated the budding of the virus. In EBOV, the budding process involved the interaction of NEDD4 with the PPxY motif of VP40, a viral matrix protein (Yasuda et al., 2003), while ubiquitination by another HECT family E3 ubiquitin ligase WWP1 was found to be important for the egress of EBOV particles (Han et al., 2017). NEDD4 was also found to be involved in the budding and release of HTLV-1 through interaction with its Gag protein (Bouamr et al., 2003).
Aside from viral exit, other viral life processes, such as escape of the virus from host endosomes, regulation of the host immunity, etc. also involve HECT ubiquitin ligases. During the entry of IAV, host E3 ligase “Itch” ubiquitinates the matrix protein M1 facilitating the release of the virus from endosomes (Su et al., 2013). The matrix protein VI of AdVs was shown to recruit NEDD4, which is required for the nuclear localization of the virus (Wodrich et al., 2010). The work of a number of research groups revealed that HPV E6 protein binds at an alpha-helical LXXLL-containing peptide motif of E6AP. This binding causes a conformational change in E6 which facilitates its interaction with p53, followed by p53 ubiquitylation and degradation (Brimer et al., 2017; Đukić et al., 2020; Scheffner et al., 1993). The UL56 protein from HSV-2 was shown to bind NEDD4. This interaction was found to involve the PY motif of UL56 and it caused phosphorylation and degradation of NEDD4. The significance of this regulation of NEDD4 during HSV-2 infection remained unclear (Ushijima et al., 2008).
RBR E3 Ligases
The RBR E3 ligases have proved to be a unique family of RING-HECT hybrid E3 ligases, which contain two RING domains, instead of one, linked by an in-between-RING domain (Dove et al., 2016; Zhang, 2008). The N-terminal RING domain acts like a RING E3 ligase and catalyzes the transfer of ubiquitin from E2 to substrates, while the C-terminal RING domain acts like HECT protein and forms an intermediate of ubiquitin linked with E3 (Zhang, 2008). These are relatively less studied for their roles in viral infection. One of the members, Triad3A, downregulates TLR, TNF-α, and RLR pathways causing suppression of antiviral responses (Nakhaei et al., 2009). Feng et al. (2004) reported an interaction of Triad 3 with the Vif protein of HIV-I, causing a reduction in the infectivity of the virus. Another RBR ubiquitin ligase ARIH1 was found to stabilize cGAS, increasing its antiviral potential against HSV-I (Xiong et al., 2022). One of the well-studied E3 ligase LUBAC was identified as the negative regulator of the TRIM25-RIG-I signaling pathway by causing proteasomal degradation of TRIM25 (Davis and Gack, 2015).
U-Box Type E3 Ligases
These E3 ligases are similar in structure as RING E3 ligases, except they do not contain the metal chelating residues of RING ligases. Like RBR ligases these are also less studied in the context of viral infections. Two of the U-box E3 ligases, CHIP and Prp19, have been discussed in the context of viral infections (Metzger et al., 2014). CHIP was found to cause ubiquitin-related degradation of the HIV-1 Tat protein, resulting in negative regulation of HIV-1 replication (Ali et al., 2019).
Table 1 presents a summary of the protein–protein direct interactions discussed in the context of all four classes of E3 ligases, along with the viral or host systems affected by these interactions.
Interactions Between Viral Proteins and Host Ubiquitin Ligases and Their Effects on the Viral Infection
AdV, adenovirus; DENV, dengue virus; EBOV, Ebola virus; EBV, Epstein–Barr virus; HCMV, human cytomegalovirus; HEV, hepatitis E virus; HPIV, human parainfluenza virus; HPV, human papillomavirus; HRSV, human respiratory syncytial virus; HTLLV‐I, human T‐cell leukemia virus type 1; IAV, influenza A virus; IDV, influenza D virus; JEV, Japanese encephalitis virus; KSHV, Kaposi’s sarcoma‐associated herpesvirus; LANA, latency‐associated nuclear antigen; RSV, respiratory syncytial virus; Rta, replication and transcription activator; TRIM, tripartite motif‐containing; ZIKV, zika virus.
Future Prospects of Antiviral Strategy Targeting Ubiquitin–Proteasome System
Majority of Viral infections are considered major health threats, both because of their high rate of host-to-host propagation and lack of treatments showing adequate success (Adamson et al., 2021; Chakrabartty et al., 2022). Although, a lot of progress has been made in early diagnosis of viral infections and considerable successes have been achieved in controlling HIV and curing other viral infections, such as HCV, lots of challenges are still associated with antiviral drug discovery (Adamson et al., 2021; Xu et al., 2022). Present antiviral therapies consist mainly of vaccines and drugs that can control viral infections by inhibiting the functions of viral proteins (also called “direct-acting antivirals”). Such therapies demand extensive research and not fast enough to counter the growing number of emerging viruses of increasing virulence (Chakravarty and Yang, 2023).
Recently, targeted protein degradation (TPDs) are emerging as an alternative antiviral strategy, in which E3 ubiquitin ligases are used as a tool for degradation of specific viral proteins. In this process, small molecules are designed to bind an E3 ligase and also viral target protein, which normally is not a substrate for the bound ligase. They may also be designed as “proteolysis targeting chimeras (PROTACs),” where the ligase is covalently attached with the ligand that binds the target protein (Bricelj et al., 2021). Ubiquitination of the target protein occurs due to their closeness with the ubiquitin ligase, subsequently resulting in the degradation of the protein (Fig. 4). So far, the most common ligases that have been used for this purpose are CRLs. Primarily, this novel treatment process has been investigated on cancer treatment, with some of the TPD products being part of clinical trials. They are predicted to play promising roles in antiviral therapy as well (Ahmad et al., 2023; Chakravarty and Yang, 2023). PROTAC-based antiviral approach was demonstrated by Si et al. (2022), who designed an attenuated IAV by tagging different viral proteins with a peptide (proteasome targeting domain, PTD) that could be recognized by a ubiquitin ligase. The tagged proteins were found to be efficiently hydrolyzed in cell lines causing replication attenuation of the engineered viruses. The degradation products of the viral proteins could induce an immune response, thus making the engineered virus a potential vaccine candidate. De Wispelaere et al. (2019) modified Telaprevir, an antiviral drug designed to bind and inhibit the nonstructural 3/4A protease of HCV. The drug was derivatized with various linkers conjugated with the ligands of a CRL. The modified drug caused both inhibition and selective ubiquitin-related degradation of the viral protease. A similar approach was taken with another antiviral drug oseltamivir, which inhibits of neuraminidases preventing exit of the newly synthesized viral particle from the cell surface (Xu et al., 2022). The above works are just a few examples of innumerable ways these heterobifunctional antiviral molecules can be designed. It has opened a whole new possibility of designing drugs that can overcome the resistance problems of the conventional antiviral drugs. Emergence of this technology has increased the importance of ubiquitin ligases, not only as targets for antiviral drugs but also as a principal part of a novel antiviral system.

Schematic presentation of the mechanism of action of PROTACs. PROTAC, proteolysis targeting chimeras.
Conclusion
In the classical approach of antiviral therapy prevention (vaccine candidates) has been considered better than cure (therapeutic drugs). After the emergency of the COVID-2019 pandemic, the current global research has been focused on understanding the viral nature for early diagnosis and therapeutic development. Viral-host protein–protein interactions provide pivotal knowledge on viral nature and characteristics, which guides better diagnosis as well as therapeutic development. During the course infection, viruses try to overcome the host defense system as well as harness the cell machinery to carry on the viral replication process. The enormous power of host ubiquitin proteins to modulate the protein stability and specificity is useful for viruses to walk a fine line in sorting out the favorable host cell proteome to complete the infection process. Ubiquitin E3 ligase, being at the foreground of this system is the ideal cell machinery to focus on, for understanding the host virus relation. Clarification of virus specificity in controlling the ubiquitin system via an appropriate interaction network is useful to identify the host factors that can be targets for designing antiviral drugs, as well as help develop novel treatment strategies for viral diseases.
Footnotes
Acknowledgment
We are thankful to Indian Council of Medical Research, Government of India for providing a Senior Research Fellowship to S.V. to carry out this work.
Authors’ Contributions
S.V.: conceptualization, data curation, project administration, validation, writing—original draft; A.G.: conceptualization, project administration, supervision, visualization, writing—review and editing.
Author Disclosure Statement
The authors have no relevant nonfinancial interests to disclose.
Funding Information
This work received no specific grant from any funding agency.