
Abstract
Interferon (IFN) is a pivotal agent against hepatitis B virus (HBV) in clinic, but there is a lack of accurate biomarkers to predict the response to IFN therapy in patients with chronic hepatitis B (CHB). Our study aimed to investigate potential targets for IFN therapy and to explore the network of interactions associated with IFN response. MicroRNA (miRNA) (GSE29911) and messenger RNA (GSE27555) datasets were used to screen the differentially expressed miRNAs (DEmiRNAs) and differentially expressed genes (DEGs). The random forest and k-nearest neighbors algorithm were used to further screen the core DEmiRNAs and build a prediction model. A Protein-Protein Interaction (PPI) network based on the STRING database was constructed and visualized by the Cytoscape software. Then, we collected transcription factors (TFs) from the TransmiR database to construct the TF-miRNA-hub gene regulatory network. Finally, real-time quantitative polymerase chain reaction was used to verify the expression of four miRNAs in HepG2-NTCP and Huh-7, and the effect of IFN treatment on four miRNAs’ expression was preliminarily explored. Eighteen DEmiRNAs in GSE29911 and 700 DEGs in GSE27555 were identified. Boruta feature selection identified four miRNAs (miR-873, miR-200a, miR-30b, and let-7g) from 18 DEmiRNAs. We identified 48 TFs, 4 miRNAs, and 10 hub genes and constructed a TF-miRNA-hub gene network to suggest the mechanism of IFN response. According to the experimental results, miR-873 was upregulated and IFN treatment could inhibit it in HBV-transfected cells (p < 0.05). We constructed a TF-miRNA-hub gene regulatory network, and our results demonstrate that miR-873 was identified as a potential biomarker of IFN response in patients with CHB. This information provides an initial basis for understanding the complex IFN response regulatory mechanisms.
Introduction
Hepatitis B virus (HBV) infection is a global public health issue, with an estimated 257 million individuals projected to be infected with HBV in 2022, reflecting a global prevalence of approximately 3.2% (Polaris Observatory Collaborators, 2023). Long-term HBV infection is the most crucial reason for the development of chronic hepatitis B (CHB). It is estimated that 20–30% of patients with CHB may further develop into cirrhosis and hepatocellular carcinoma, which are the main causes of hepatitis B-related mortality (GBD 2013 Mortality and Causes of Death Collaborators, 2015; Hsu et al., 2023; Schweitzer et al., 2015). Thus, early and effective antiviral therapy is essential to slow down the progression of CHB. Currently, interferon (IFN) and nucleos(t)ide analogues (NAs) are the primary treatments available for CHB (Suresh and Menne, 2022). Similarly, peginterferon (PEG-IFN) is employed in the treatment of patients with CHB. In comparison to NAs, PEG-IFN has been demonstrated to have the advantages of shorter treatment time, lower drug resistance rate, and stronger immune regulatory effect (European Association for the Study of the Liver, 2017; Terrault et al., 2018). However, PEG-IFN treatment is commonly accompanied by adverse events including influenza-like symptoms, neutropenia, thrombocytopenia, and so forth (Li et al., 2024). The traditional biomarkers, including HBV DNA, HBsAg, and ALT levels, have been used to predict IFN treatment response, but their accuracy is still limited (Sonneveld et al., 2013; Zhang et al., 2020); Buster et al., 2009), and the emergence of novel biomarkers is urgently needed.
MicroRNAs (miRNAs) are a group of small endogenous single-stranded noncoding RNA molecules that act as key regulators in a variety of biological processes, including differentiation, proliferation, apoptosis, invasion, stress response, and immunity (Bhayani et al., 2012; Giovannetti et al., 2012). miRNA has attracted much attention as an emerging biomarker, and it has been shown that HBV-miR-3 is a new biomarker of HBV replication and an early predictor of HBeAg seroconversion in patients with CHB treated with Peg IFN-α (Xu et al., 2024). Additionally, exosomal miRNAs (miR-194-5p and miR-22-3p) screened by next-generation sequencing were identified to predict HBeAg seroconversion in patients with CHB treated with Peg-IFN (Hu et al., 2021). Nevertheless, the current body of research on miRNA as a predictor of IFN therapy response remains limited, and further studies are required to identify miRNAs with potential as biomarkers.
In this study, we obtained a dataset of miRNAs and messenger RNAs (mRNAs) associated with the IFN response in patients with CHB from the GEO database. We constructed a transcription factor (TF)-miRNA-hub gene coexpression network to identify potential biomarkers of the IFN response. Subsequently, we preliminarily validated the effect of IFN treatment on miRNAs’ expression through cell experiments.
Materials and Methods
Data collection
Series matrix files of GSE29911 (Zhang et al., 2012) and GSE27555 were downloaded from the GEO (http://www.ncbi.nlm.nih.gov/geo/) database. The platforms they based on were GPL10406 (Agilent-021827 Human miRNA Microarray Rel12.0, v3.0, 8x15k array) and GPL6480 (Agilent-014850 Whole Human Genome Microarray 4x44K G4112F), respectively. The dataset of GSE29911 contained 94 plasma samples from patients with CHB receiving IFN therapy, of which 40 were responders to IFN therapy and 54 were nonresponders to IFN therapy. The datasets of GSE27555 included liver biopsy specimens from seven patients who did not respond to IFN therapy and six patients who responded.
Identification of differentially expressed miRNAs and differentially expressed genes
The limma package of R (version 3.4.0, https://www.r-project.org/) was applied for differential analysis to screen for differentially expressed miRNAs (DEmiRNAs) and differentially expressed genes (DEGs). p-Value <0.05 and | log2(Fold Change) | ≥2 were set as the threshold values in DEmiRNAs, and p-value <0.05 and | log2(Fold Change) | ≥1 were considered cutoff criteria in DEGs.
Variable selection and prediction model construction
Feature selection of DEmiRNAs was performed using the Boruta package based on the random forest algorithm (Kursa and Rudnicki, 2010). Compared with other selection-by-variable method, the Boruta method had fairly low out-of-bag error rates and computation times. Boruta tests if the importance of each individual variable is significantly higher than the importance of a random variable by fitting random forest models iteratively until all predictor variables are classified as confirmed or rejected at the 0.05 alpha level. In the present study, 94 patients with CHB receiving IFN treatment were randomly assigned to training and test groups. The training set consisted of 29 IFN treatment responders and 36 IFN treatment nonresponders. The validation set included 11 IFN treatment responders and 18 IFN treatment nonresponders. Then, a predictive model was constructed using a k-nearest neighbor (KNN) classifier to predict the outcome of IFN response in patients with CHB. As a powerful algorithm, the KNN depends upon a neighborhood of close (or similar) patterns relative to a query pattern, and an important challenge is to find the best distance or similarity measure. Therefore, we used the weighted nearest neighbor method based on KNN algorithm to obtain a better prediction model. To compare the effect of different weighting methods on the model results, we used “triangular,” “epanechnikov,” “inv” methods, and the standard “rectangular” method for analysis.
Identification and enrichment analyses of 106 mRNAs
By using starBase v2.0 online tool, the interactions between the mRNAs and miRNAs were obtained. Seven databases (PITA, RNA22, miRmap, microT, miRanda, PicTar, and TargetScan) about predicted miRNA and mRNA binding can be linked to starBase v2.0. Only interactions between miRNAs and mRNAs that can be retrieved from more than two databases were retained. Then, we screened the common mRNAs between the target mRNAs of DEmiRNAs and the DEGs. To explore the potential biological function of common mRNAs, Gene Ontology (GO) functional annotation, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, and Reactome pathway enrichment analysis were performed by using the “clusterProfiler” package of R language. p-Value <0.05 was considered as statistically significant.
Construction of the TF-miRNA-hub gene regulatory network
Interactions between common mRNAs were obtained by using the STRING database, a database that provides information on direct (physical) and indirect (functional) associations between different proteins. In order to discover genes that play a key role in the PPI network, the cytoHubba module of the Cytoscape v3.7.2 software was used to identify hub genes in the PPI network. The cytoHubba module assigns values to each gene through a variety of topology network algorithms (Degree, EPC, MNC, DMNC, MCC), ranking the values according to the size of the algorithm assignment to discover hub genes and subnetworks in the network. The TransmiR database (http://www.cuilab.cn/transmir) was used to predict TF-miRNA relations. Based on the regulation relationship between TFs, miRNAs, and hub genes, the TF-miRNA-hub gene network was constructed and visualized using Cytoscape v3.7.2 software.
Cell culture and treatments
The HepG2-NTCP and Huh-7 were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin–streptomycin. For transfection studies, HBV genome containing 1.1 copies was transfected into cells using Lipofectamine 2000 (Invitrogen, USA) according to the instructions of the reagent. Cells were treated with recombinant human IFN-α-2b injection (1,000 IU/mL) from Anke Biotechnology (Anhui, China) to simulate IFN treatment in patients. After the transfected cells were cultured in 37°C incubator for 48 h, the supernatant and cells were extracted.
Real-time quantitative polymerase chain reaction
Total RNA was extracted from treated cells using RNAi plus (TaKaRa, Japan). Complementary DNA (cDNA) of miRNA was synthesized using 2×miRNA P-RT Solution Mix and miRNA P-RT Enzyme Mix (Sangon Biotech., China). Quantitative polymerase chain reaction (PCR) was performed using SYBR Green qPCR Master Mix Kit (MedChemExpress, China) on the BioRad® 96 instrument Real-Time PCR System. The used primer sets are presented in Table 1. The expression of miRNA was calculated using 2−△△Ct method and was normalized to that of U6.
Sequences of miRNA Primers
Results
Identification of DEmiRNAs and DEGs
In the present study, 18 DEmiRNAs in GSE29911 and 700 DEGs in GSE27555 were identified. Among the DEmiRNAs and DEGs, 3 and 448 genes were upregulated while 15 and 252 genes were downregulated in GSE29911 and GSE27555, respectively. The differentially expressed DEmiRNAs and DEGs were visualized by volcano plots (Fig. 1). Red dots represent the upregulated genes, and green dots represent the downregulated genes. Then, we sorted by | log2FC | and annotated the genes in the front row. The results showed that DEmiRNAs were mostly downregulated and miR-873 upregulated in IFN response group.

The differentially expressed miRNAs and genes were visualized by volcano plots.
Screening of core DEmiRNAs based on machine learning and constructing prediction models
Boruta feature selection identified four miRNAs (miR-873, miR-200a, miR-30b, and let-7g) from 18 DEmiRNAs for constructing prediction models (Fig. 2). To obtain the best classification performance, we used triangular, epanechnikov, inv, and rectangular for KNN fitting, and the random seed number was 22. It was found that the highest prediction accuracy was achieved when the weighting method was “inv” and K = 10. The classification error in the training set was 0.2307692, the classification accuracy was 76.92%, and the prediction accuracy in the test set was 72.41%.
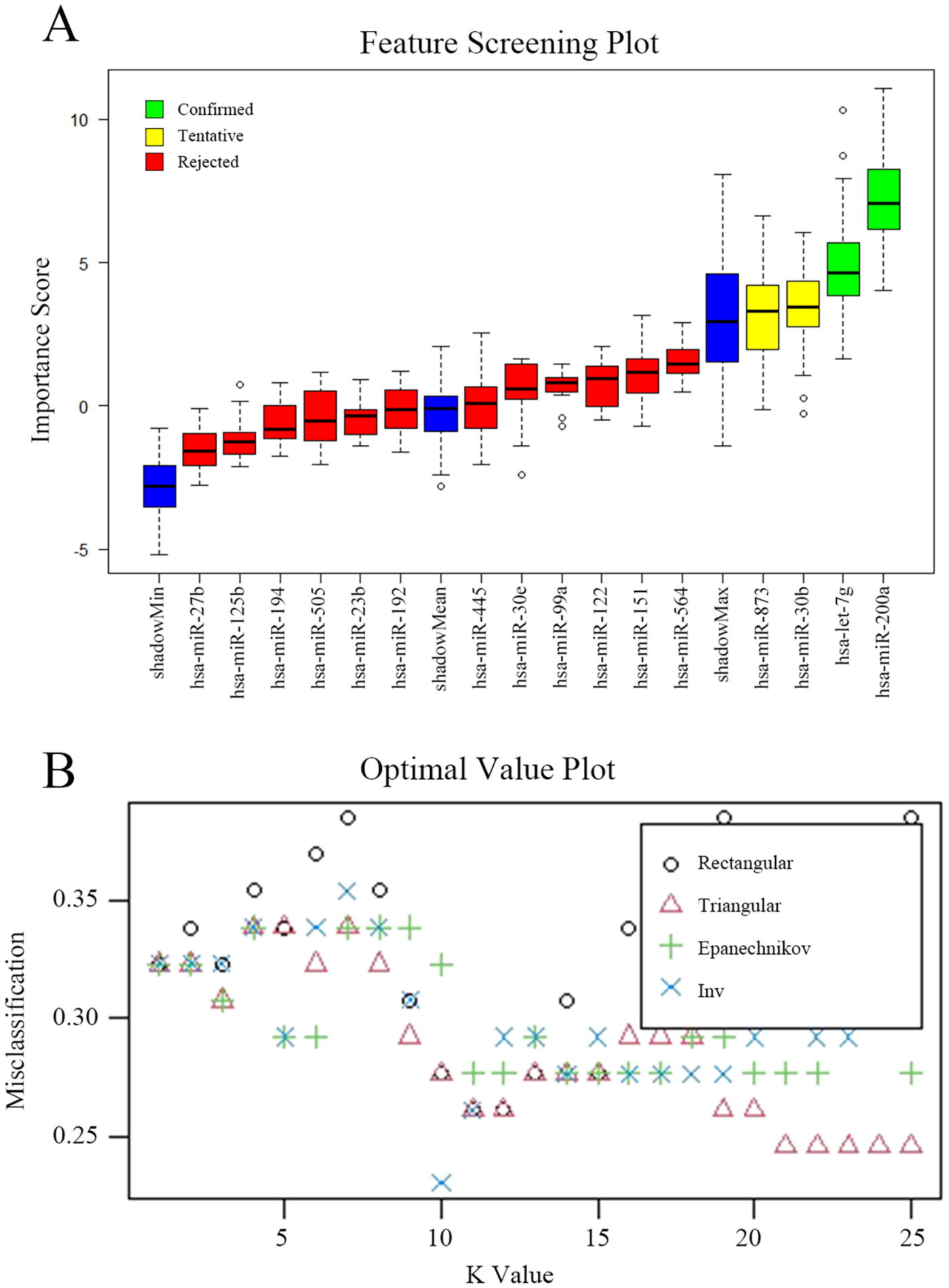
Boruta feature selection identified miR-873, miR-200a, miR-30b, and let-7g for constructing prediction models.
Identification and enrichment analyses of common mRNAs
We obtained 5,204 target genes of four miRNAs by target gene prediction. Next, we obtained 106 mRNAs (Fig. 3A) by intersecting the 5,098 target genes of four miRNAs with the previously obtained 700 DEGs. Finally, 106 mRNAs were used to explore the potential functions of miRNA, including GO enrichment analysis, KEGG pathway analysis, and Reactome pathway analysis. In the GO analysis, we obtained 219 results. The top 10 clusters with their representative enriched terms of biological process are shown in Figure 3B. The 106 mRNAs were mainly involved in the regulation of biological processes related to Leukocyte transendothelial migration, such as actin filament organization, T-cell activation, and cell adhesion. KEGG analysis showed that 106 mRNAs were mainly enriched in six pathways. The common mRNAs were mainly enriched in HBV-related pathways such as PI3K-AKT, JAK-STAT, and other cancer-related pathways (Fig. 3C). The reactome pathways analysis of 106 mRNAs were shown to be enriched in the cytokine signaling in immune system, signaling by interleukins (Fig. 3D).
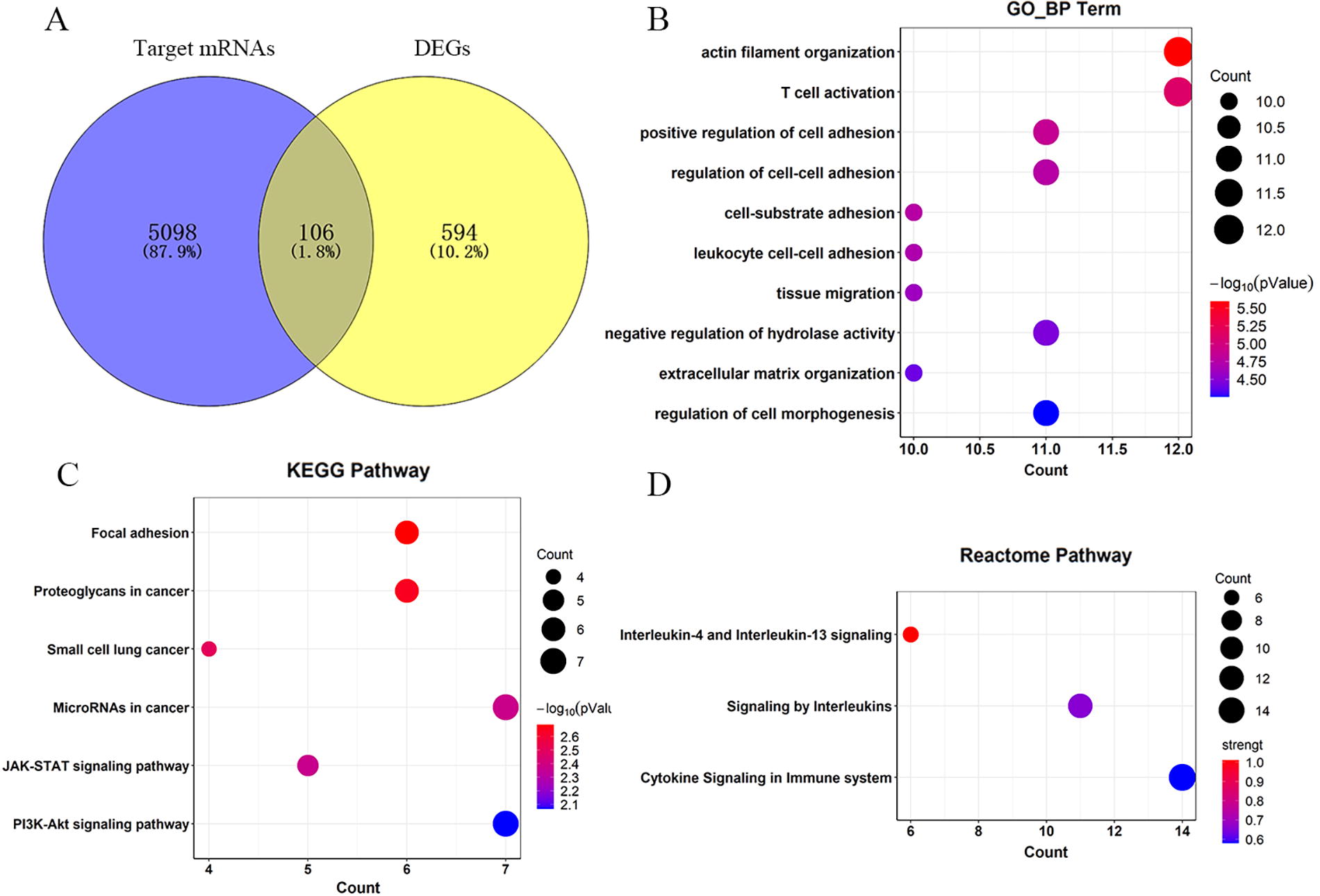
Identification and enrichment analyses of common mRNAs.
Construction of the TF-miRNA-hub gene regulatory network
To further find the hub genes of 106 common mRNAs in the network, we analyzed the PPI network with STRING database and Cytoscape (Fig. 4A). And we identified 10 hub genes showing higher degrees of connectivity using the cytoHubba plug-in (Fig. 4B). These hub genes included ACTA1, IQGAP1, ACTG2, GSN, ANXA2, FLNA, CD44, FGF2, MMP2, and MKI67. As shown in Figure 4C, we constructed a TF-miRNA-hub gene network to present potential regulatory mechanisms. The network consists of 62 nodes and 115 edges, including 48 TFs, 4 miRNAs, and 10 key genes.
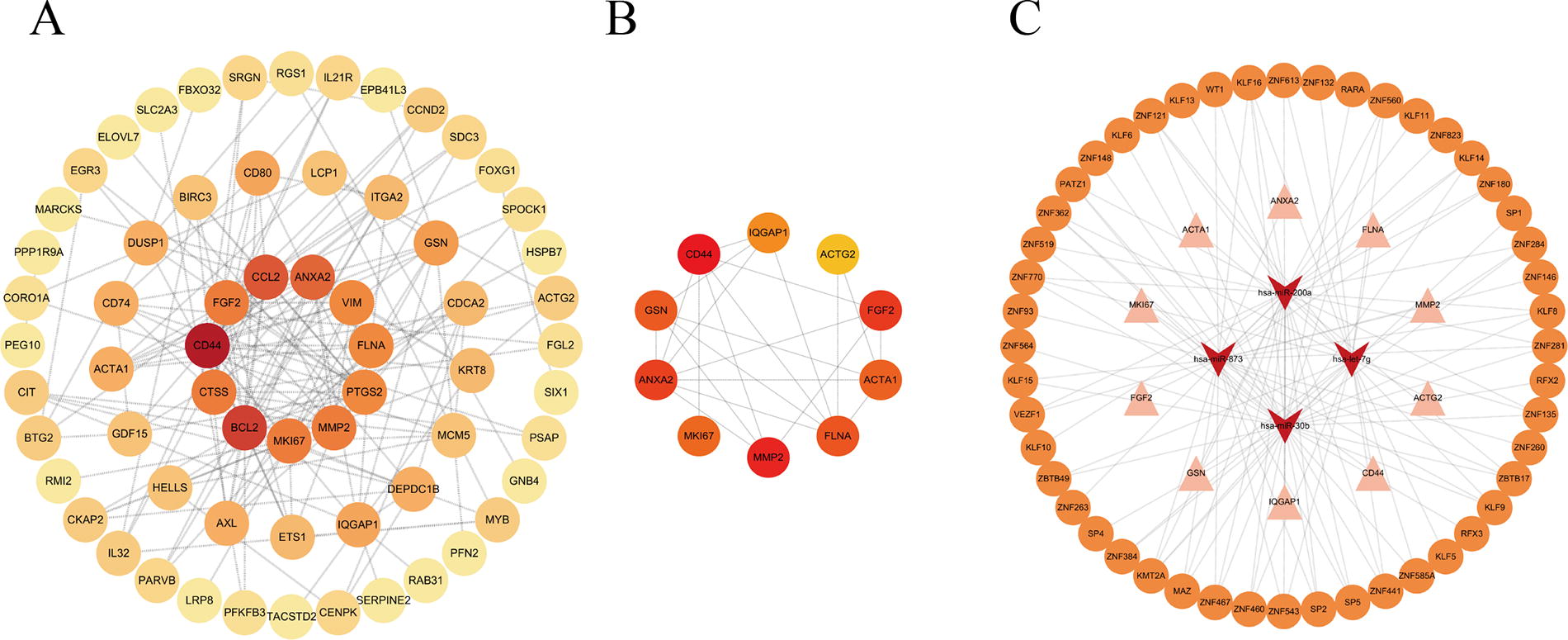
Construction of the PPI and TF-miRNA-hub gene network with STRING database and Cytoscape.
Validation of featured DEmiRNAs expression in Huh-7 and HepG2-NTCP cells
In this study, Huh-7 and HepG2-NTCP cells were selected for testing of whether DEmiRNAs were subject to regulation by HBV infection. We analyzed miRNAs expression in HBV plasmid transfection of Huh-7 and HepG2-NTCP cells by real-time quantitative PCR analyses. As shown in Figure 5A, the expression of miR-873 was significantly upregulated in Huh-7 cells and HepG2-NTCP cells (p < 0.001). Moreover, our results indicated that miR-200a, miR-30b, and let-7g were also upregulated in Huh-7 cells (p < 0.05) and HepG2-NTCP cells (p < 0.01), respectively (Fig. 5B–D).
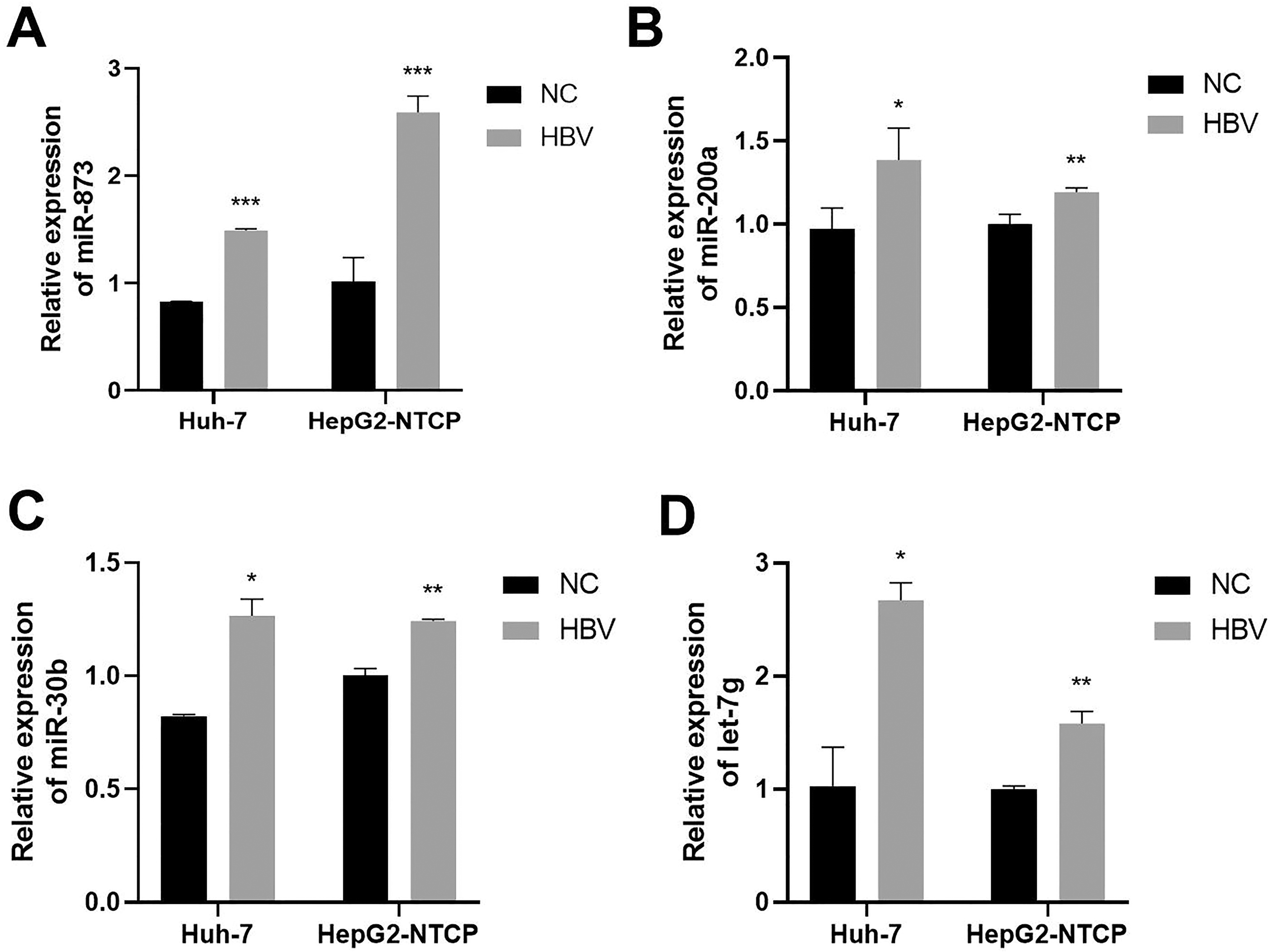
The differential expression of
IFN-α induces downregulation of miR-873
Huh-7 and HepG2-NTCP cells transfected with the HBV replication plasmid pHBV1.1 were treated with 1,000 U/mL of IFN-α to investigate the expression of miRNAs. The results showed that the expression level of miR-873 was obviously decreased in IFN-treated cells (p < 0.05, Fig. 6A). The expression level of miR-200a only in HepG2-NTCP cells treated with IFN was significantly reduced (p < 0.01, Fig. 6B), while there was no significant difference in miR-30b level between IFN treated and untreated cells (p > 0.05, Fig. 6C). The expression level of let-7g was significantly reduced only in Huh-7 cells treated with IFN (p < 0.05, Fig. 6D). Therefore, miR-873 may most likely play a role in the process of HBV infection and the mechanism of IFN action.
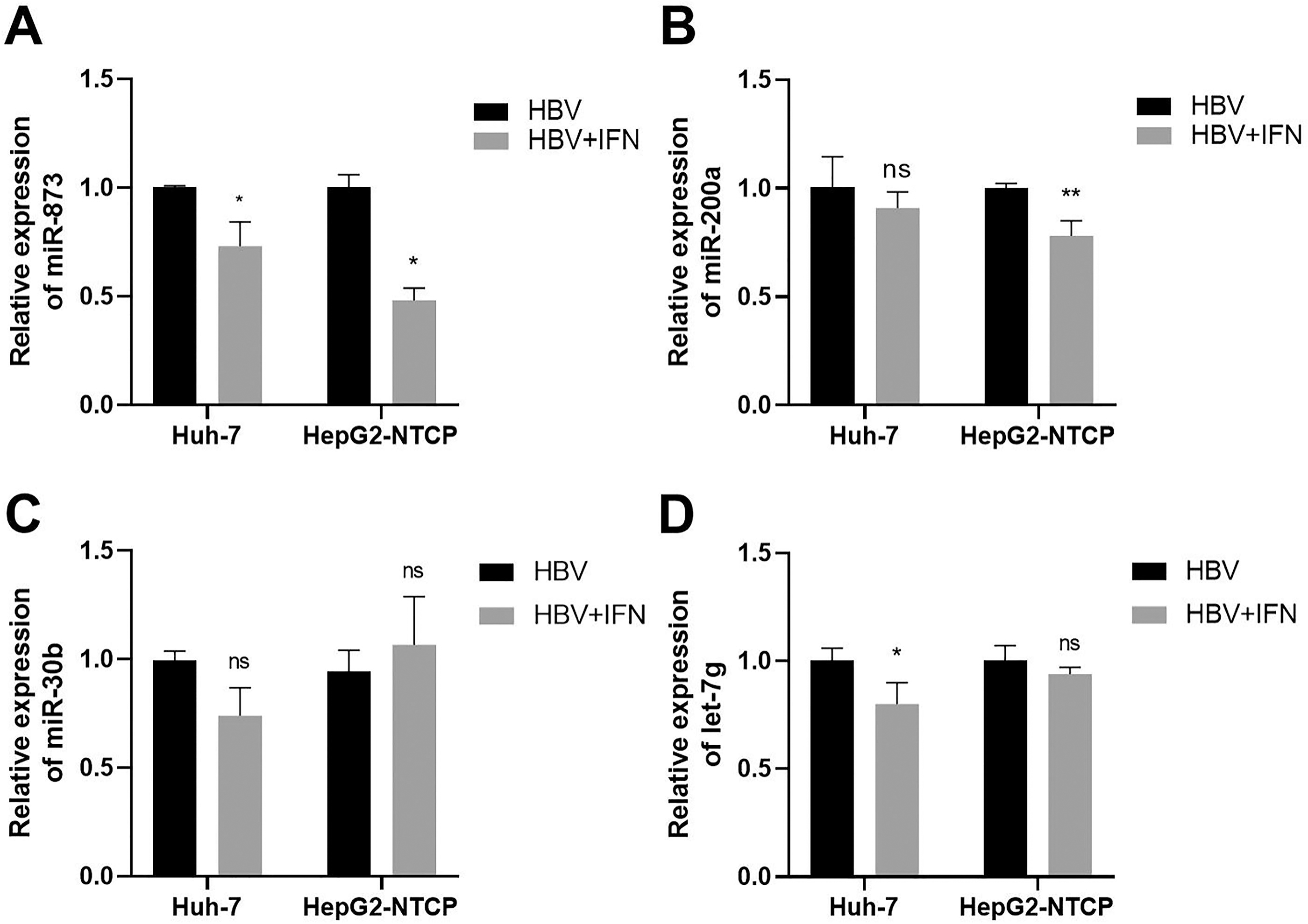
Effect of HBV infection and IFN treatment on miRNAs expression in Huh-7 and HepG2-NTCP cells
Discussion
CHB represents a significant public health concern, with a substantial disease burden on humans. IFN is one of the first-line therapeutic options for patients with CHB, but the clinical use of IFN is constrained by side effects and low response rates (European Association for the Study of the Liver, 2017; Kwon and Lok, 2011; Terrault et al., 2018). Hence, in order to improve the overall success rate of IFN therapy, it is indispensable to distinguish between possible responders and unlikely responders before the initiation of IFN therapy. In this study, we obtained a TF-miRNA-hub gene network involved in IFN regulation and investigated the effect of IFN treatment on the expression levels of four DEmiRNAs (miR-873, miR-200a, miR-30b, and let-7g), which could provide new references for anti-HBV therapy.
We obtained 18 DEmiRNAs and 700 DEGs associated with IFN responses using the GEO dataset. Then, we used random forest and KNN algorithms to identify four core miRNAs, miR-873, miR-200a, miR-30b, and let-7g, that are more closely associated with IFN responses. In addition, a prediction model for IFN response was established using these four core miRNAs, with an accuracy of 76%. This further demonstrates the correlation between core miRNAs and IFN responses. Compared with the study of GSE29911 (Zhang et al., 2012), they used a support vector machine algorithm to construct a model consisting of 11 miRNAs variables for predicting IFN response with a prediction accuracy of 71.4%. Our study constructed a model with higher prediction accuracy using fewer predictors by differential expression analysis and Boruta algorithm. In several hepatocellular carcinoma–related studies, HBV infection affects the expression levels of miR-200a and let-7g in tumor tissues (Du et al., 2022; Shi et al., 2017; Wakasugi et al., 2018). HBV infection promotes miR-30b expression through the interaction of HBV protein P with FOXO3 (Chen et al., 2021).
With the aim of exploring the biological functions of core miRNAs, we performed an intersection of 700 DEGs with the target mRNAs of core miRNAs to obtain the most potential 106 common mRNAs associated with IFN therapy. Next, GO term, KEGG pathway, and Reactome pathway enrichment analysis were performed on common mRNAs. The GO analysis results indicate that common mRNAs play important roles in actin filament organization, T-cell activation, and cell adhesion. These biological processes are important components of leukocyte transendothelial migration. As known, leukocyte transendothelial migration is one of the most essential steps in inflammatory response and immune defense (Kolaczkowska and Kubes, 2013; Muller, 2011). Besides, Reactome pathway analysis showed that common mRNAs are involved in the regulation of cytokine and inflammatory factor signaling such as interleukin-4 (IL-4). Interestingly, miR-873 could promote the differentiation of Th17 cells (Liu et al., 2017). IL-17 produced by Th17 cells induces miR-873 to promote the production of inflammatory cytokines (Liu et al., 2014). Thus, it is worth investigating whether miR-873 exerts its immunosuppressive effect by affecting leukocyte migration. According to the result of KEGG pathway enrichment analysis, common mRNAs are mainly enriched in hepatitis B-related pathways and cancer pathways. Multiple studies have demonstrated that the PI3K-AKT pathways play essential roles in resistance to HBV replication and the development of hepatocellular carcinoma (Meng et al., 2014). And miR-873 was found to inhibit PI3K/AKT pathway activation (Yang et al., 2021). These results reveal that miR-873, miR-200a, miR-30b, and let-7g may be involved in the regulatory and functional pathways of IFN therapy response.
To identify the regulatory network associated with IFN response, we constructed a TF-miRNA-hub gene network consisting of 48 TFs, 4 miRNAs, and 10 hub genes. In this regulatory network, IQGAP1 is a highly conserved scaffolding protein, and it has been suggested that IQGAP1 may regulate type I IFN production by interacting with NLRC3 in human monocytes and epithelial cells (Tocker et al., 2017). ANXA2 (Annexin A2), a phospholipid-binding protein, has extensive tissue distribution and biological functions. Cumulative evidence confirms the interaction of IFN-γ and ANXA2, with IFN-γ promoting the secretion of ANXA2 and ANXA2 regulating IFN-γ-induced autophagy (Chen et al., 2017). TFs are DNA-binding proteins that play a vital role in the regulation of gene expression, and TFs can regulate the expression of the HBV genome and affect viral replication and pathogenicity (Ren et al., 2012). Previous studies have shown that the transcription factor KLF15 (Krüppel-like factor 15) is a novel transcriptional activator of the HBV core and surface promoters, and KLf15 has the potential to be a potential therapeutic target for reducing HBV gene expression and viral replication (Zhou et al., 2011). Moreover, ZNF148 (Zinc finger protein 148) inhibits HBV replication by downregulating RXRα transcription, suggesting that ZNF148 may be a target for a novel anti-HBV strategy (Yao et al., 2024). To the best of our knowledge, this is the first attempt to integrate patients with CHB’s IFN response-associated miRNA and mRNA expression profiling data and construct a TF-miRNA-hub gene regulatory network. The identification and analysis of IFN response-related genes, miRNAs, and TFs may reveal potential mechanisms of IFN response in patients with CHB.
Finally, we verified the expression of the four miRNAs in HBV-transfected cells and further observed the changes in the expression levels of the four miRNAs after treatment with IFN. The results showed that miR-873 was upregulated in HBV-transfected cells and downregulated in IFN-treated cells. Nevertheless, no significant differences were observed simultaneously in miR-200a, miR-30b, and let-7g in the two HBV-transfected cell models after IFN treatment. Clearly, our study uncovers valuable IFN-related candidate targets, miR-873, that may serve as biomarkers of response to IFN therapy.
There are several limitations of this study that should be acknowledged. First, we constructed a predictive model of whether patients with CHB responded to IFN therapy, and the predictive efficacy of the model was unsatisfactory despite the use of more stringent variable screening criteria and more appropriate algorithms. However, compared with the study based on this miRNA dataset, the model we constructed was somewhat more advantageous and was able to achieve higher predictive accuracy with fewer detection indicators. Second, we only preliminarily explored the effect of IFN treatment on the expression of the four miRNAs, and more in vivo and in vitro experiments are needed in the future to validate their relationship with IFN treatment.
Conclusions
A TF-miRNA-hub gene regulatory network was constructed through bioinformatics analysis and machine learning of mRNA and miRNA expression data. After experimental validation, it was found that miR-873 is the most probable biomarker for IFN therapy response. These findings contribute to a more comprehensive understanding of the underlying molecular mechanisms and provide potential target biomarkers for IFN response in patients with CHB.
Footnotes
Authors’ Contributions
X.Y.: Conceptualization, methodology, writing—original draft, and validation. M.Z.: Conceptualization, methodology, and validation. X.L.: Data curation, formal analysis, and visualization. J.F.: Resources and writing—review and editing. L.C.: Validation. J.L. and S.L.: Data curation. L.Z.: Conceptualization, funding acquisition, resources, supervision, and writing—review and editing. All authors have contributed significantly, and all authors agree with the content of the article.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Author Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Information
This work was supported by the Program for Youth Innovation in Future Medicine, Chongqing Medical University (Grant No. W0177) and the Natural Science Foundation of Chongqing (Grant No. CSTB2024NSCQ-MSX0169).