
Abstract
Cell senescence, induced by various internal and external stresses, plays a significant role in the development of various diseases such as cancer, neurodegeneration, and infections. Viral infections can also induce cellular senescence, known as virus-induced senescence (VIS), which occurs in close correlation with the severity of the viral infections. However, due to the unclear mechanisms underlying VIS, the effective inhibition of VIS during viral infections is challenging, leading to rapid disease progression. This study utilized the widely used vesicular stomatitis virus (VSV) model virus to simulate RNA virus infections for exploring the mechanisms by which RNA viruses induce cellular senescence. The results indicated that VSV infection, both in vitro and in vivo, could significantly induce the upregulation of senescence-associated markers and the secretion of the senescence-associated secretory phenotype (SASP), promoting the senescence process. Further research found that the activation of the NF-κB pathway played a crucial role in VSV-induced cellular senescence. Targeted inhibition of the NF-κB pathway could reduce the level of organ senescence induced by viral infections, decrease the expression of SASP inflammatory factors, and ameliorate tissue damage in mice. Overall, our findings reveal the mechanisms underlying RNA virus-associated VIS and provide potential targets for inhibiting the occurrence of VIS and preventing disease progression.
Introduction
As age progresses, the accumulation of senescent cells increases, leading to a decline in the body’s self-regulation capabilities and an increased susceptibility to diseases (López-Otín et al., 2023). The occurrence of cellular senescence, while considered beneficial for tissue repair and immune surveillance, has also been found to be closely related to the development of various diseases, such as cancer, neurodegeneration, and infections (Di Micco et al., 2021; Gulen et al., 2023; Kelley et al., 2020; Wang et al., 2024).
Cellular senescence is defined as the permanent state of cell cycle arrest in normally proliferating cells under various internal and external stimuli, characterized by several features, including an increase in senescence-associated β-galactosidase (SA-β-gal) activity, the accumulation of cyclin-dependent kinase inhibitors such as p16 and p21, and the secretion of the senescence-associated secretory phenotype (SASP), which includes numerous cytokines, chemokines, and enzymes involved in the remodeling of the extracellular matrix, such as IL-6 and IL-1β (Di Micco et al., 2021; Gorgoulis et al., 2019; Wang et al., 2024). In addition to replicative senescence due to repetitive cell division, cellular senescence phenotypes with similar characteristics can be triggered by various cellular stresses, such as radiation, chemotherapy drugs, and pathogen infections (Di Micco et al., 2021; Gorgoulis et al., 2019). In particular, viral infections can also induce cellular senescence, known as virus-induced senescence (VIS), which has been confirmed in various viral infections, including SARS-CoV-2, influenza virus, respiratory syncytial virus, etc. (Lee et al., 2021; Li et al., 2023; Martínez et al., 2016; Schulz et al., 2023). Studies have shown that the occurrence of VIS is closely related to the progression and prognosis of viral infectious diseases, and targeting the removal of senescent cells or the inhibition of pathways or molecules related to the secretion of SASP can alleviate the severity of viral infections (Camell et al., 2021; Lee et al., 2021; Li et al., 2023; Schmitt et al., 2023). However, due to the unclear mechanisms underlying VIS, it is challenging to effectively inhibit the onset of VIS during viral infections, leading to rapid disease progression. Therefore, elucidating the mechanisms of VIS occurrence is crucial for early targeted intervention to prevent disease deterioration.
Upon viral infection, virus-related nucleic acid substances can be recognized by various pattern recognition receptors of the host cell, such as TLR3, RIG-I, and cGAS, which mediate downstream signal transduction, leading to innate immune responses and the production of inflammatory factors (Choi et al., 2018). Studies have demonstrated that the secretion of inflammatory factors plays a crucial role in promoting the process of cellular senescence (López-Otín et al., 2023). The activation of inflammatory pathways during viral infection critically depends on the type of viral nucleic acid, although the replication cycles and pathogenic mechanisms of different viruses may vary (Carty et al., 2021; Choi et al., 2018). Therefore, we hypothesize that viruses of the same nucleic acid type, that is, DNA or RNA viruses, may induce senescence in an inflammation-dependent manner through similar inflammatory pathways. Given that RNA viruses such as SARS-CoV-2 and influenza virus infect a large number of people, have a wide spread, and are more likely to cause severe diseases (Flerlage et al., 2021; Liang, 2023; Onder et al., 2020), this study chose the vesicular stomatitis virus (VSV), an RNA model virus, to simulate RNA virus infections (Ogino et al., 2024; Yang et al., 2024; Zhou et al., 2022) and explore the mechanisms by which RNA virus infections induce cellular senescence, providing potential targets for inhibiting the occurrence of VIS.
Materials and Methods
Cell culture and treatment
A549 cells were purchased from the American Type Culture Collection. Mouse embryonic fibroblast (MEF) cells were prepared from embryos at approximately 15 days of gestation. Both A549 and MEF cells were cultured in Dulbecco’s modified Eagle’s medium (GIBCO) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a humidified atmosphere of 5% CO2.
For the VSV-induced senescence experiments in vitro, VSV (multiplicity of infection = 0.1) was used to infect A549 and MEF cells. The culture medium was changed 24 h post-infection, and the cells were continued to be cultured for 5 days before sample collection and analysis. For the BAY-11-7082 treatment experiments, BAY-11-7082 (10 μM, TargetMol, T1902) was added to the cells 72 h after VSV-induced senescence, and the culture was also continued for 5 days before sample collection and analysis.
Mice and treatment
C57BL/6 wild-type mice were purchased from the Hubei Province Laboratory Animal Research Center (Wuhan, China) and were housed under specific pathogen-free conditions at the College of Life Sciences, Wuhan University. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of State Key Laboratory of Virology, College of Life Sciences, Wuhan University (approval code: SKLV-AE2023 027; Date: Jan 1, 2023), and were conducted in accordance with the Animal Welfare Act and the National Institutes of Health guidelines for the care and use of laboratory animals in biomedical research.
For the VSV-induced senescence experiments in vivo, C57BL/6 wild-type mice (8–10 weeks old) were infected with VSV via intraperitoneal injection on day 0 (1 × 107 PFU per mouse) and were euthanized for tissue collection and analysis on day 7 or 14 post-infection. For the BAY-11-7082 treatment, mice were infected with VSV on day 0, and the treatment group received BAY-11-7082 (10 mg/kg) via intraperitoneal injection on days 2, 4, and 6, while the control group received an equal volume of vehicle (corn oil and dimethyl sulfoxide). The mice were euthanized on day 7 for tissue collection and analysis.
Real-time quantitative polymerase chain reaction
Cell samples were lysed using Trizol reagent (Invitrogen), and total RNA was extracted using chloroform and isopropanol, followed by cDNA synthesis using HiScript II Q RT SuperMix (Vazyme, R222-01). Real-time quantitative polymerase chain reaction (RT-qPCR) was performed using ArtiCanCEO SYBR qPCR Mix (Tsingke, TSE401) and specific primers with cDNA as the template. The relative expression of mRNA was calculated using the 2−ΔΔCt method, and the expression levels of the target genes were normalized to a baseline sample. All primers used in this study are listed in Table 1.
Primer Sequences for qPCR
SA-β-gal staining
SA-β-gal staining was performed in VSV-infected cells at 5 days or in infected mice at 7 or 14 days, following the instructions provided in the SA-β-gal staining kit (Beyotime, C0602). Briefly, cells or tissue cryosections were washed with PBS and fixed with β-galactosidase staining fixative at room temperature for 15 min, then incubated with staining solution at 37°C overnight. The next day, images were captured and processed using a Leica inverted microscope system.
BrdU assay
5-BrdU (MedChemExpress, HY-15910) was applied to the cells at 20 μM for 24 h. After removing the culture medium, the cells were sequentially fixed with pre-cooled methanol for 15 min, permeabilized with 0.1% Triton X-100 for 10 min, and denatured with 2N HCl for 30 min, followed by blocking with 5% BSA for 1 h. The BrdU primary antibody (Proteintech, 66241-1-Ig) was added and incubated overnight at 4°C. Cy3 fluorescent secondary antibody (Abbkine, A22210) was incubated on the following day, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) before image acquisition using the Leica inverted microscope system.
ELISA
The levels of IL-6 protein in the cell supernatant were detected and analyzed according to the IL-6 (MABTECH, 3361-1H-20) enzyme-linked immunosorbent assay (ELISA) kit instructions. Briefly, capture mAb was added to a 96-well ELISA plate and incubated overnight at 4°C. The next day, the plate was blocked with incubation buffer (PBS + 0.05% Tween 20 + 0.1% BSA) for 1 h at room temperature, washed five times with wash buffer (PBS + 0.05% Tween 20), and then samples and standards were added and incubated for 2 h at room temperature. After five washes, the detection mAb was added and incubated for 1 h at room temperature. After five washes, streptavidin-HRP was added and incubated for 1 h at room temperature. Washed five times, then added 3,3′,5,5′-tetramethylbenzidine (TMB) substrate and incubated for 15 min in the dark before the reaction was stopped with 0.2M H2SO4, and the optical density was measured at 450 nm.
Histological analysis and immunohistochemistry
Histopathological damage and fibrotic changes were detected by hematoxylin and eosin (H&E) and Masson’s trichrome staining with acidic fuchsin and aniline blue, respectively. The levels of tissue p21 and IL-6 protein expression were measured utilizing immunohistochemistry. For tissue samples undergoing immunohistochemistry, tissues were fixed, dehydrated, and paraffin-embedded, and then sectioned and deparaffinized in water. Sections were then incubated in ethylenediaminetetraacetic acid (EDTA) antigen retrieval buffer and heated for antigen retrieval. After incubation in 3% hydrogen peroxide solution at room temperature for 25 min to block endogenous peroxidase, 3% BSA was added and incubated for 30 min at room temperature. After removing the blocking solution, PBS-diluted primary antibodies for p21 (Servicebio, GB12153) and IL-6 (Servicebio, GB11117) were added and incubated overnight at 4°C. Sections were washed three times with PBS and then incubated with corresponding species-specific HRP-labeled secondary antibodies for 50 min at room temperature. After three washes with PBS, DAB chromogen was added for color development, and the color development time was controlled under a microscope, followed by rinsing and stopping the staining with the sections. Harris hematoxylin was used to counterstain cell nuclei for about 3 min, followed by differentiation with 1% hydrochloric alcohol for a few seconds and ammonia water for blueing. After rinsing and dehydration, the sections were mounted, and images were observed and captured using an inverted microscope system.
Statistics and data analysis
Quantification for SA-β-gal staining and histological analysis was performed using ImageJ to determine the percentage of positively stained areas. For the results of SA-β-gal staining in cells or tissues, three to four representative samples were randomly selected for quantification. For the quantification of tissue immunohistochemistry and Masson staining results, four representative samples were quantified. Quantitative results for each group were normalized to show relative values using the control group as a reference. All data were analyzed using at least three comparable independent experiments and are expressed as mean ± SD. GraphPad Prism software (v9.5.1) was used for statistical analysis. Two-tailed Student’s t-tests were performed for comparisons between two groups, and one-way analysis of variance, followed by Tukey’s multiple comparison test was used for multiple-group comparisons. A p-value less than 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).
Results
VSV induces senescence in vitro
We initially infected A549 cells (human non-small cell lung cancer cells) with VSV and observed a significant increase in SA-β-gal activity (Fig. 1A, B), along with markedly elevated levels of senescence-associated markers such as p16 and p21, and SASP inflammatory factors such as IL-6 and IL-1β (Fig. 1C) after 5 days. To determine the universality in which VSV infection in vitro can stably induce cellular senescence, we repeated the above experiments using MEF cells. Similarly, after VSV infection of MEF cells for 5 days, there was a significant upregulation of senescence-associated markers, SASP inflammatory factor levels, and SA-β-gal activity (Fig. 1D–F). Meanwhile, to further characterize the cell cycle arrest of senescent cells, we examined cell proliferation through the BrdU assay. The results showed that VSV infection of MEF cells for 5 days significantly reduced cell proliferation activity (Fig. 1G), indicating that the cells had entered a state of cell cycle arrest. Therefore, these results indicate that VSV, as an RNA model virus, can stably induce cellular senescence upon in vitro infection.
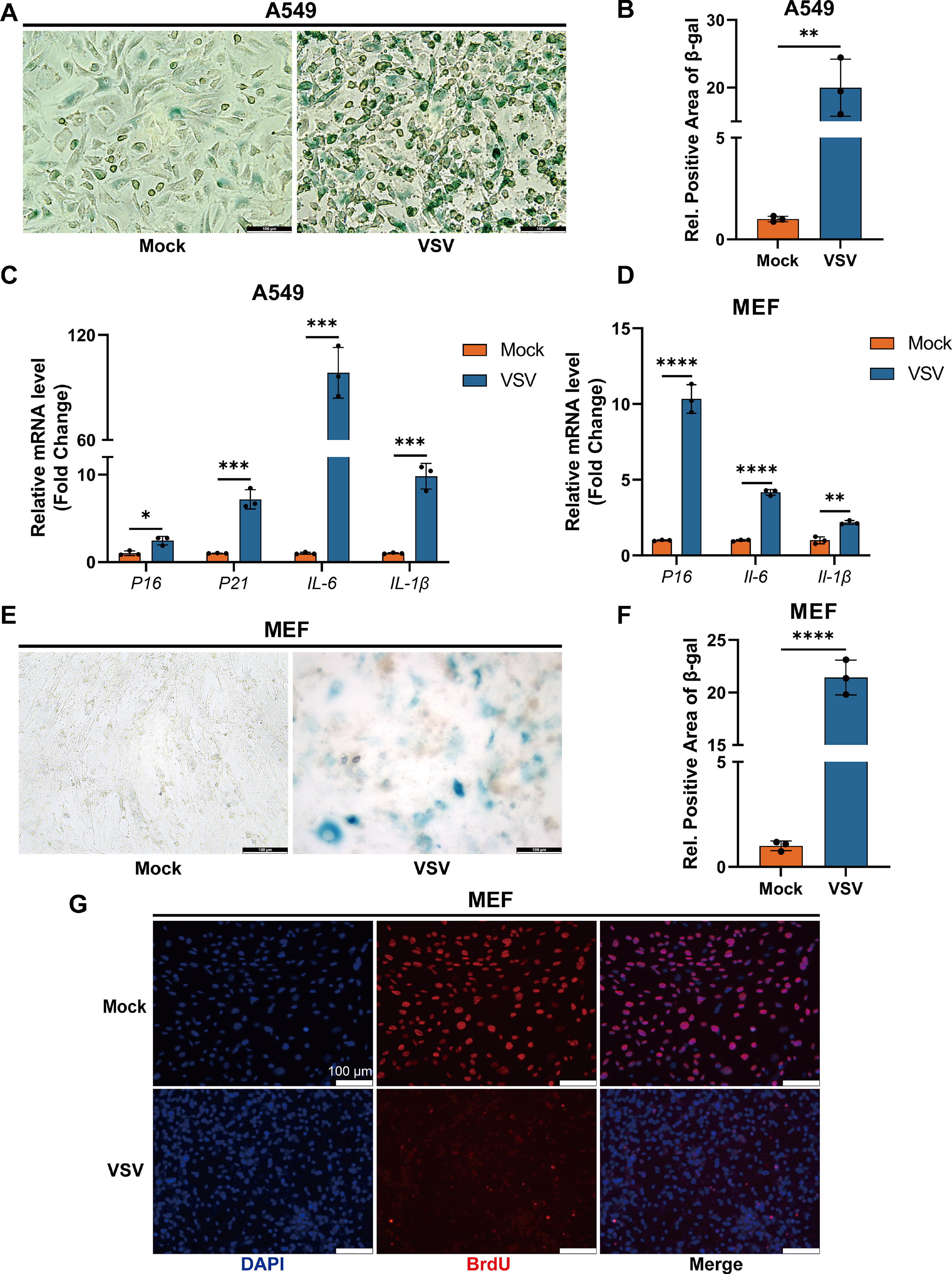
VSV induces senescence in vitro.
VSV induces organ senescence and persistent inflammation injury in vivo
To further explore whether VSV infection could induce senescence in vivo, we infected C57BL/6 wild-type mice with VSV via intraperitoneal injection for 7 or 14 days. The results showed that, compared to the control group, there was a significant deposition of SA-β-gal in the lungs and livers of mice infected with VSV for 7 days, and this phenomenon persisted at 14 days post-infection, particularly in the liver (Fig. 2A, B). Correspondingly, immunohistochemical results revealed a significant increase in the expression levels of senescence and inflammation markers such as p21 and IL-6 (Fig. 2C–F). Additionally, H&E and Masson staining indicated that VSV infection also led to damage and fibrosis in the lungs and livers of mice that persisted to 14 days post-infection (Fig. 2G–J). Therefore, these experimental results demonstrate that VSV infection in vivo can induce significant organ senescence, with the liver being the most affected, accompanied by persistent inflammation and tissue injury.

VSV induces organ senescence and persistent inflammation injury in vivo.
NF-κB pathway activation mediates VSV-induced cellular senescence
Our previous studies have proven that VSV infection, both in vivo and in vitro, can induce cellular senescence, promote the senescence process, and lead to persistent inflammation and injury. Since inflammation and cellular senescence are usually considered to have a mutually enhancing relationship, and various pathogens, such as bacteria and viruses, often induce an inflammatory response through the activation of the NF-κB pathway during infection (López-Otín et al., 2023; Sun, 2017). Therefore, we further explored whether the mechanism of VSV-induced cellular senescence is related to the activation of the NF-κB pathway. We used the small molecule drug BAY-11-7082 to target the activation of NF-κB (Guruvaiah and Gupta, 2024; Mori et al., 2002) in vitro and found that inhibiting NF-κB during VSV-induced senescence of A549 cells significantly reduced the deposition of SA-β-gal (Fig. 3A, B), and lowered the levels of senescence markers and SASP inflammatory factors such as p21 and IL-6 (Fig. 3C). We further repeated the experiment in MEF cells and similarly found that inhibiting NF-κB could significantly reduce the expression levels of p21 and IL-6 (Fig. 3D, E), as well as SA-β-gal activity (Fig. 3F, G), slowing down the cellular senescence process. Meanwhile, the BrdU assay results also demonstrated that inhibiting NF-κB alleviated VSV-induced cell cycle arrest in senescent cells (Fig. 3H). These results indicate that the activation of the NF-κB pathway mediates the VSV-induced cellular senescence process, and targeted inhibition of NF-κB can significantly reduce the levels of cellular senescence and inflammation.

NF-κB pathway activation mediates VSV-induced cellular senescence.
Targeting the NF-κB pathway in vivo inhibits the VSV infection-related senescence process and persistent inflammation injury
Our previous results in vitro found that VSV-induced cellular senescence is mediated by the NF-κB pathway, so we proceeded to further validate this in vivo. We first infected mice with VSV via intraperitoneal injection on day 0 and then treated the experimental group with BAY-11-7082 medication on days 2, 4, and 6, while the control group received an equal volume of vehicle treatment. Samples were collected for analysis on day 7 post-infection (Fig. 4A). The results showed that, compared to the control group, the levels of the senescence marker p21 in the lungs and livers of the experimental group mice were significantly reduced (Fig. 4B, C), and the expression levels of the inflammatory factor IL-6 were also markedly inhibited (Fig. 4D, E). Additionally, treatment with BAY-11-7082 significantly improved tissue damage and fibrosis in the lungs and livers (Fig. 4F–I), indicating that by inhibiting NF-κB pathway-mediated cellular senescence, one can alleviate the persistent inflammation and tissue damage associated with VSV infection. Therefore, the above experimental results demonstrate that VSV induces organ senescence and persistent inflammation and injury in vivo through the NF-κB pathway, and targeting NF-κB has a certain therapeutic effect.
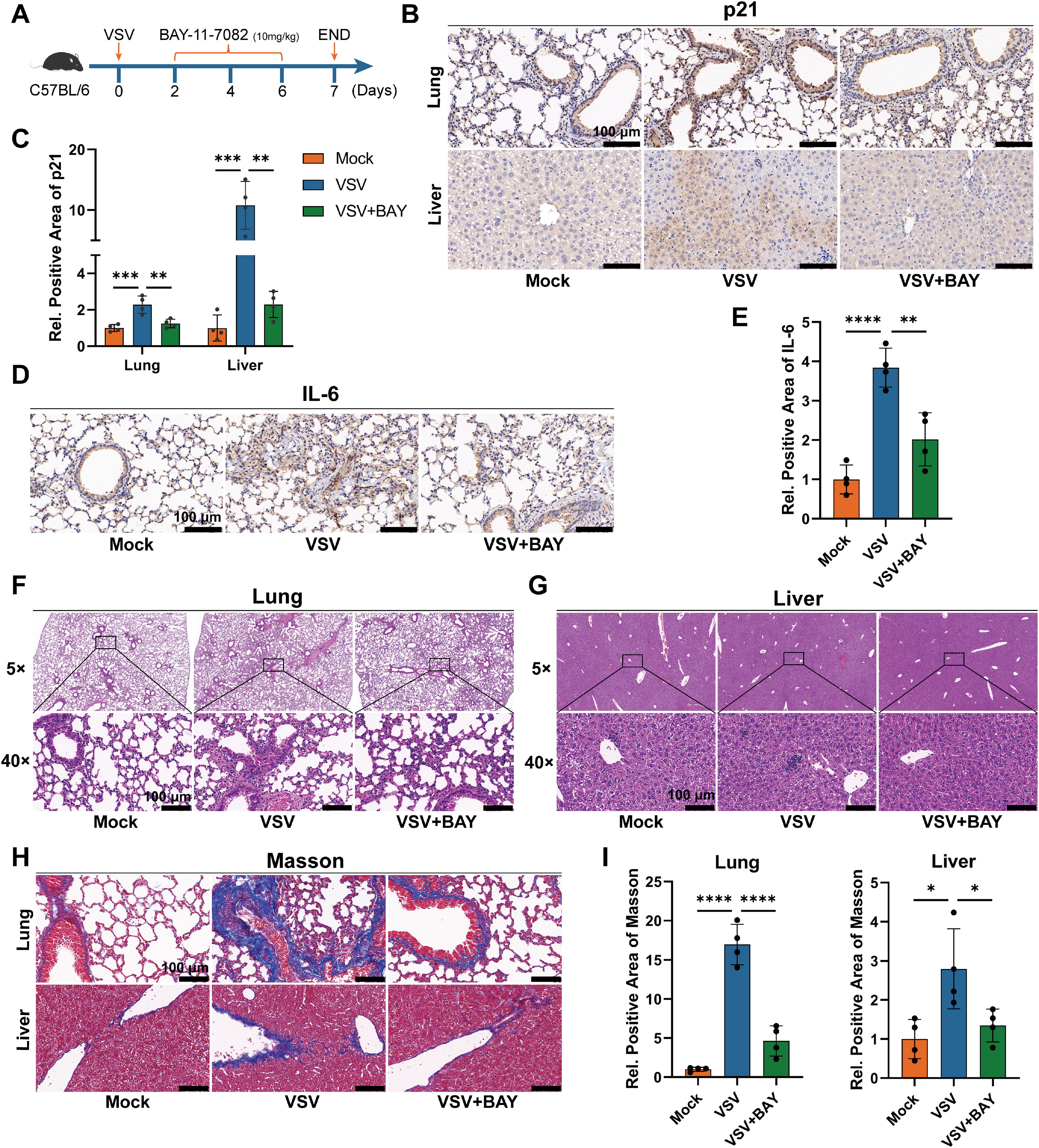
Targeting the NF-κB pathway in vivo inhibits the VSV infection-related senescence process and persistent inflammation injury.
Discussion
VSV is a non-segmented single-stranded negative-sense RNA virus and a representative species of the family Rhabdoviridae. VSV is widely used as a model virus for studying viral pathogenesis, such as the characteristics of antiviral innate immune response and viral replication, owing to its simple structure, strong replication ability, and limited pathogenicity, especially for RNA viruses such as influenza virus, SARS-CoV-2, rabies virus, and Ebola virus (Ogino et al., 2024; Ogino and Green, 2019; Poeck et al., 2010; Yang et al., 2024; Zhou et al., 2022). Thus, the study used VSV to simulate RNA virus infection to explore the potential mechanisms by which RNA virus infection induces cellular senescence in an inflammation-dependent manner through innate immune pathways. Our results proved that VSV can promote the senescence process both in vivo and in vitro by activating the NF-κB pathway, and the accumulation of senescent cells correlates closely with persistent inflammation and damage related to viral infection. Targeted inhibition of NF-κB pathway activation can reduce the level of senescence induced by viral infection and slow down disease progression.
It has been widely reported that viral infections can induce cellular senescence (Lee et al., 2021; Li et al., 2023; Schulz et al., 2023), but the specific mechanisms of VIS are still unclear, making it very difficult to intervene in VIS in advance and prevent VIS from promoting disease deterioration. Several studies have revealed the mechanisms by which specific viral infections such as measles virus (Chuprin et al., 2013), hepatitis B virus (Idrissi et al., 2016), human immunodeficiency virus (Beaupere et al., 2015; Chauvin and Sauce, 2022), cytomegalovirus (Wu et al., 2023), herpes simplex virus (Sivasubramanian et al., 2022), influenza A virus (Schulz et al., 2023; Yan et al., 2017), and SARS-CoV-2 (Schmitt et al., 2023) induce cellular senescence, mainly including the activation of the p53 and p16-pRb pathways (Chuprin et al., 2013; Idrissi et al., 2016; Sivasubramanian et al., 2022; Wu et al., 2023), DNA damage response induced by mitochondrial oxidative stress (Gioia et al., 2023; Martínez et al., 2016; Schmitt et al., 2023; Wu et al., 2023), and the regulation of specific viral proteins (Beaupere et al., 2015; Gioia et al., 2023; Idrissi et al., 2016; Noris et al., 2002; Yan et al., 2017). Notably, our study found that VSV induces cellular senescence by activating the NF-κB pathway, providing new insights for further understanding and targeting the occurrence of VIS related to RNA viruses.
Existing research suggests that there is a mutually enhancing relationship between inflammatory responses and cellular senescence, that is, chronic inflammatory stimulation can trigger cellular senescence, and senescent cells can continuously secrete SASP-related inflammatory factors (López-Otín et al., 2023; Tsuji et al., 2022). NF-κB, as a nuclear transcription factor, plays an important role in various biological processes such as immune responses, inflammation regulation, and cell growth (Sun, 2017; Yu et al., 2020). Studies have shown that the activation of the NF-κB pathway is also closely related to aging, and targeting NF-κB has been proven to inhibit inflammation and age-related tissue degeneration (Chien et al., 2011; Josephson et al., 2019; Yoon et al., 2022; Yu et al., 2020). However, before this, the role of the NF-κB pathway in VIS was not clear, especially for RNA viruses with strong pathogenicity and widespread, such as the influenza virus, SARS-CoV-2, and Ebola virus. Therefore, using VSV as a model virus, we reveal the key role of NF-κB in RNA virus-induced cellular senescence, which is of great significance. However, although we employed the widely used and recognized VSV model virus to simulate RNA virus infections to explore the mechanisms of senescence induction, there is a lack of related experimental data using other specific viruses directly. Therefore, although the conclusions of this study are enlightening for the process of RNA virus-induced senescence, whether other specific types of RNA viruses have the same mechanism still needs further research.
Although we have tried our best to provide higher-quality experimental results, this study still has certain limitations. First, the A549 cells used in our in vitro model are a lung adenocarcinoma cell line, which may complicate the senescence-associated pathways involved. Although we also employed a VSV-induced senescence model using primary MEF cells, potential discrepancies with alternative models should still be considered. Second, in our exploration of the mechanism of VSV-induced cellular senescence, we only studied the NF-κB pathway, and the process may also involve other pathways or specific regulatory molecules, which still need further analysis and experimentation.
Footnotes
Authors’ Contributions
Z.L., M.T., and C.Z. conceived the study. Z.L., H.F., J.X., M.T., C.Y., S.L., and G.L. designed, performed, and analyzed experiments and interpreted data. C.Z., K.W., and J.A.G.S. supervised the experiments. Z.L., H.F., and J.X. drafted the article. K.W. and C.Z. contributed to the editing of the paper. All authors read and approved the final version of the article.
Data Availability Statement
All data are available in the main text.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This work was supported by the National Key R&D Program of China (2023YFC2308404), the National Natural Science Foundation of China (32188101, 32400131, 82472337), National Key Research and Development Program of China (2023YFC2306600), China Postdoctoral Science Foundation (2024T170687, 2024M752482, GZB20230541) and the Open Research Program of the State Key Laboratory of Virology of China (2023KF004, 2022KF003).