
Abstract
Human metapneumovirus (HMPV) is a prominent respiratory pathogen causing significant morbidity and mortality worldwide, mostly in young teenagers, the old, and immunocompromised individuals. Despite its clinical impact, no licensed vaccine is currently available, highlighting the urgent need for effective prophylactic strategies. This research aimed to design a multiepitope vaccine (MEV) targeting conserved and immunodominant regions of HMPV, leveraging immunoinformatics tools to ensure broad coverage and efficacy against the virus and its diverse sublineages. Glycoproteins from HMPV genotypes A2a, A2b, and A2c were analyzed to identify 18 highly antigenic and overlapping epitopes capable of eliciting robust B-cell, T-cell, and interferon-gamma (IFN-γ)-mediated immune responses. Toxicity and allergenicity studies confirmed the safety of particular epitopes, which were incorporated into two vaccine constructs using immunogenic linkers and adjuvants. The chimeric vaccines displayed high antigenicity, molecular stability, and nonallergenic properties. Structural refinement and Ramachandran plot analyses established the stability and accuracy of the 3D models. Molecular docking studies verified strong interactions with immune receptors, particularly toll-like receptor (TLR)2, TLR3, TLR4, TLR8, and human leukocyte antigen molecules, indicating robust immune stimulation potential. Molecular dynamics simulations further validated the vaccine’s stability and interaction dynamics, with immune simulations predicting promising responses. The designed vaccine constructs were shown to be highly soluble, stable, and suitable for recombinant expression in Escherichia coli, enabling further biochemical and immunoreactivity validation. These findings provide a foundation for next-generation vaccine development against HMPV, offering promising avenues for clinical application and future research.

Introduction
Human metapneumovirus (HMPV), first discovered in 2001 among 28 children in the Netherlands, has emerged as an important contributor to acute respiratory tract infections, particularly affecting children, the elderly, and immunocompromised individuals (Deffrasnes et al., 2007). Typically, HMPV circulates seasonally from winter to spring alongside other respiratory viruses, causing a limited number of cases. Between 2016 and 2021, the average detection rate among individuals under 18 was approximately 4% (Kahn, 2006). However, an unprecedented surge occurred in 2023, with hospitals and clinical laboratories across the United States reporting a 19.6% positivity rate for HMPV antigen tests in March—substantially higher than the 3.6% average recorded from 2008 to 2014 (Bhasin et al., 2024). This spike led to significant strain on pediatric and intensive care facilities (Fig. 1).
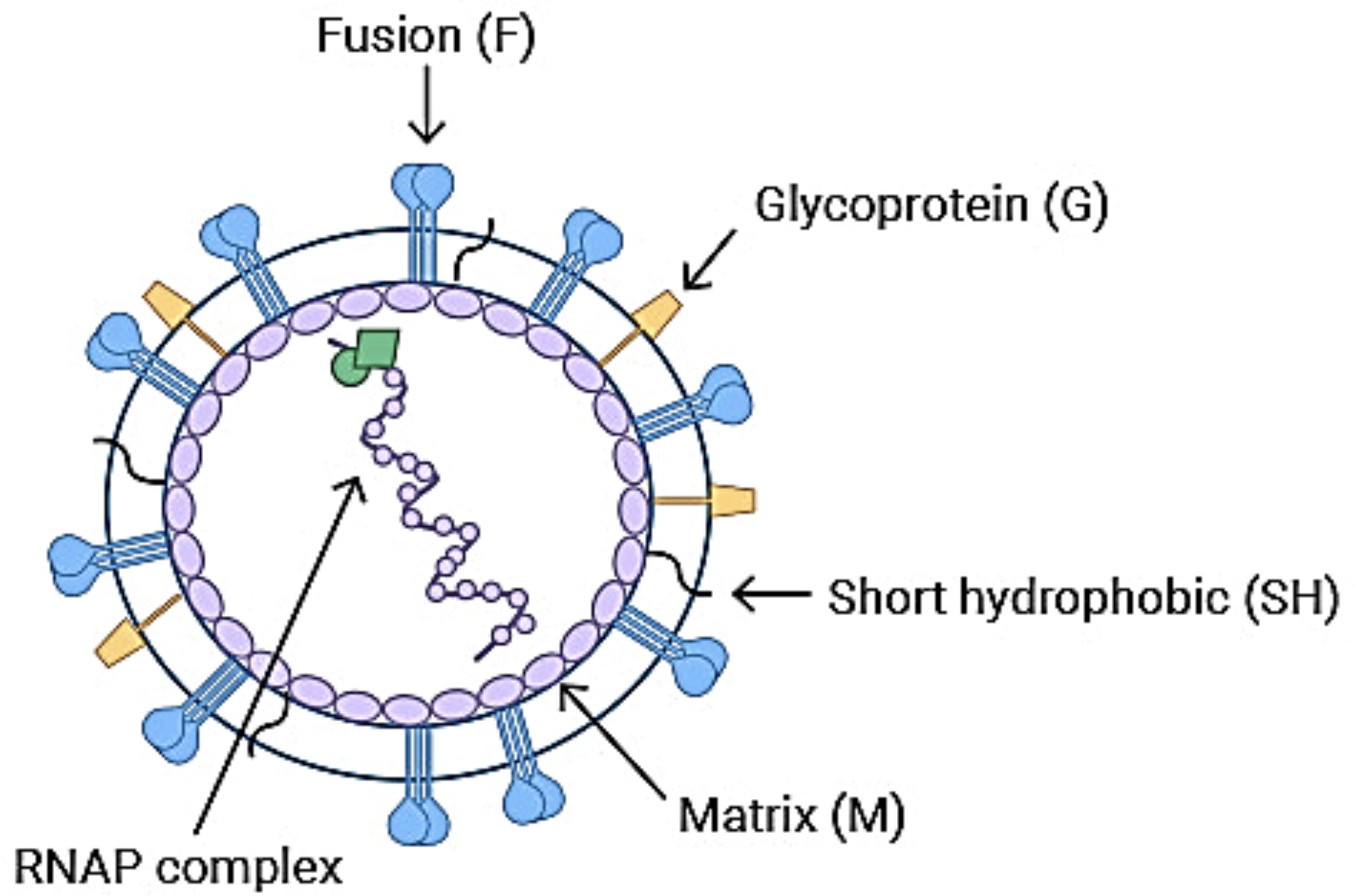
Diagram of human metapneumovirus (HMPV) with components.
HMPV is a member of the Pneumoviridae family, shares a clinical profile with respiratory syncytial virus and accounts for approximately 15% of acute respiratory infections in young children and 5% in adults (Walsh et al., 2008). The World Health Organization reported that HMPV contributed to 10% of deaths among children under five in 2017. In 2018, an estimated 14 million children under five were infected globally, with 640,000 requiring hospitalization and 8,000 fatalities recorded (Ma et al., 2024). While HMPV typically causes mild symptoms such as fever, cough, and nasal congestion, individuals with weakened immune systems, including premature infants, may develop severe complications such as bronchiolitis, pneumonia, or life-threatening respiratory distress necessitating intensive care (Esposito and Mastrolia, 2016). The virus imposes a substantial health care burden annually, particularly in resource-limited regions. Despite the urgent need for effective interventions to protect vulnerable populations, no vaccine or definitive treatment currently exists. This gap contributes to significant hospitalization rates and mortality during outbreaks (Lefebvre et al., 2016).
Therapeutic approaches for HMPV infection currently rely on supportive interventions such as immunoglobulins, glucocorticoids, and symptomatic treatments. In severe cases, clinical application includes ribavirin and polyclonal intravenous immune globulin, although robust evidence from randomized controlled trials validating their efficacy is lacking (De Zwart et al., 2022). These treatments are implemented based on clinical experience rather than conclusive data. Experimental strategies with antiviral potential, such as monoclonal antibodies, RNA interference (RNAi), NMSO3, and heparin, have shown promise in preclinical models, including studies in mice. However, none of these have yet received regulatory approval for clinical use (Kumar and Srivastava, 2018).
Advancing vaccine development is a promising strategy to mitigate the burden of viral infections, particularly in the absence of effective therapies (Panda et al., 2014). HMPV has diversified into two primary genetic lineages, A and B, each containing two subtypes: A1, A2, B1, and B2 (Devanathan et al., 2025). Subtype A2 is further categorized into A2a, A2b, and A2c. Despite genetic differences, these variants share a high level of protein sequence similarity, with over 80% homology observed across subtypes. This conservation highlights the pressing need for a comprehensive vaccine capable of addressing the dynamic variants of HMPV, particularly those within the A2 subgroup (Mizuta et al., 2010).
Utilizing a range of immunoinformatics approaches, this study developed and assessed a multiepitope vaccine (MEV) targeting HMPV. Glycoprotein sequences were retrieved from the National Center for Biotechnology Information (NCBI), and antigenicity was evaluated using VaxiJen. Epitope prediction for B-cells, cytotoxic T-cells (CTL), and helper T lymphocyte (HTL) was conducted via the Immune Epitope Database (IEDB) server, while epitope-receptor interactions were analyzed through molecular docking using ClusPro and HADDOCK. The vaccine construct incorporated epitopes linked with adjuvants, optimized using linker tools, and its stability was examined through molecular dynamics (MDs) simulations in GROMACS. Codon optimization and in silico cloning for Escherichia coli expression were performed using the JCat server. This integrative strategy enhances immune responses by combining multiple viral epitopes, minimizing the likelihood of escape variants, and providing a versatile solution for combating HMPV.
Materials and Methods
Retrieval protein sequences
Three genotypes of the HMPV virus (A2a, A2b, and A2c) were retrieved from the NCBI database (Pruitt et al., 2005). Among these, glycoproteins were chosen for their pivotal role in host–pathogen interactions, including receptor binding and immune system evasion, making them prime targets for immune responses. Their intricate structure, composed of protein and carbohydrate elements, stimulates robust and diverse immune reactions, such as antibody production and T-cell activation. Furthermore, the conserved nature of glycoproteins across various strains enhances their potential for broad-spectrum immunity (Hariharan and Kane, 2020; Lembo et al., 2024). Their surface localization also facilitates accessibility to immune mechanisms, highlighting their utility in designing efficient MEVs. For each HMPV variant, two glycoproteins (accession numbers: BAV89108.1, BAV89107.1, BDE17504.1, BAV89161.1, BES52865.1, and BES52863.1) were selected to predict immunogenic epitopes.
Prediction of T-cell epitopes
T-cell epitopes were identified using the IEDB server, focusing on conserved sequences (Dhanda et al., 2019). For cytotoxic T lymphocytes (CTL), the epitope length was predefined as 9 amino acids, a standard size that ensures effective binding to Major Histocompatibility Complex (MHC) class I molecules by using Artificial Neural Network (ANN) 4.0 method. Following this, a length of 12 amino acids was selected for HTL epitope prediction, as this size facilitates interaction with MHC class II molecules by utilizing NN-align 2.3 (NetMHCII 2.3) method with (IC50 < 100 nM), essential for their role in aiding other immune cells. The epitope generation process targeted both CTL and HTL, incorporating MHC class I alleles (human leukocyte antigen [HLA] A, B, C, and E) and class II alleles (HLA DRB).
Prediction of B-cell epitopes
The IEDB B-cell epitope analyzing tool was utilized to predict potential B-cell epitopes through various methodologies, which assesses protein regions for accessibility and flexibility—important traits of functional B-cell epitopes. Furthermore, epitope prediction was supplemented using the ABCpred server, configured with its standard settings, to identify epitopes within the target proteins (Malik et al., 2022). Linear epitopes were identified using BepiPred-3.0 (threshold: 0.5), while conformational epitopes were predicted with DiscoTope 2.0 (threshold: −3.7) and ElliPro (≥70% percentile rank).
Validation of nominated B-cell and T-cell epitopes
The epitopes were selected based on comprehensive evaluations of antigenicity, allergenicity, topology, and toxicity, utilizing VaxiJen v2.0 (threshold: 0.4) (Doytchinova and Flower, 2007), AllerTOP v2.0 (Dimitrov et al., 2014), TMHMM (Sorathiya et al., 2020), and ToxinPred v3.0 servers (Rathore et al., 2023). These parameters were employed to identify candidates with optimal attributes for vaccine development. Antigenicity was assessed via VaxiJen v2.0 with a 0.4 threshold, while AllerTOP excluded potential allergens using default criteria. Topological properties were analyzed with TMHMM v.2.0 to confirm surface accessibility, and ToxinPred eliminated toxic candidates. Epitope lengths were standardized: 9 amino acids for CTL, 12 for HTL, and 12–16 for B-cell epitopes, ensuring immunogenicity and structural compatibility. The Support Vector Machine (SVM) method predicted IFN-γ-inducing epitopes critical for innate and adaptive immunity using the IFNepitope tool, while IL10 pred was employed to assess IL-10-inducing epitopes, ensuring a balanced immune response (Kak et al., 2018). This approach ensures that the selected epitopes not only enhance protective Th1 immunity but also contribute to immune regulation, minimizing potential immunopathology. The rigorous selection process guarantees that the epitopes exhibit safety, immunogenicity, and functional relevance for vaccine formulation.
MEV design and validation
The most antigenic, nonallergenic, and nontoxic 9–16 mer CTL, HTL, and linear B-cell epitopes were nominated for the design of chimeric vaccine models. The overlapping epitopes were chosen to elicit both cell-mediated and robustic immune responses targeting highly antigenic epitopes. To further enhance immune activation and promote long-term innate immunity, potential immune-stimulatory adjuvants were incorporated. Two adjuvants, Heparin-Binding Hemagglutinin Adhesin (HBHA) protein and β-defensin, were integrated into the design (Semple et al., 2010). The EAAAK linker was used to link the adjuvant to the N-terminus, while flexible linkers, GGGS and HEYGAEALERG, facilitated effective epitope conjugation following Solanki and Tiwari’s methodology (Solanki et al., 2019). Additionally, PADRE enhances vaccine immunogenicity and stability by providing universal T-helper epitopes, ensuring robust and sustained immune responses (Didelon et al., 2020; Del Guercio et al., 1997). The chosen linkers prevent unwanted junctional epitopes, boost the expression and bioactivity of the construct, and promote strong immunogenicity.
The allergenicity of the designed vaccines was evaluated utilizing the AllerTOP v2.0 server. To assess antigenicity and solubility, the VaxiJen v2.0, ToxinPred, and Protein-Sol servers were employed, facilitating the identification of the most promising candidate. Additionally, the ExPASy website provided insights into the physiochemical properties of the vaccine model. This comprehensive approach allowed for a thorough assessment, increasing the likelihood of selecting a safe and effective vaccine for further development. For predicting longstanding protection, immunoinformatics approaches such as IEDB’s Class I and II MHC Binding Prediction and MEMORY were used to examine memory B-cell and T-cell epitopes (Sanchez-Trincado et al., 2017). The approaches expedited the evaluation of the chosen epitopes ability to trigger robust immune responses, crucial for long-lasting defense against HMPV.
2D and 3D structures’ prediction and assessment
The PSIPRED online website, known for its high accuracy in secondary structure analysis, was used to forecast the alpha-helices, beta-sheets, and coil regions of the MEVs (Buchan and Jones, 2019). Its precision was carefully validated through cross-validation to assess prediction reliability. The I-TASSER algorithms were then applied to analyze the tertiary structure of the MEVs (Zhou et al., 2022). To further refine the 3D model, the GalaxyRefine tool is used. The excellence of the polished structure was subsequently evaluated using a Ramachandran plot, produced by the PROCHECK web server (Laskowski et al., 1993). Additionally, the VERIFY 3D and ERRAT websites were applied to examine the accuracy of the predicted tertiary structure.
Molecular docking
Toll-like receptors (TLRs) are pivotal in detecting harmful pathogens and initiating inflammatory responses (Zhu et al., 2023). Among these, TLR8 plays a significant role in the immune system’s reaction to infectious agents (Cervantes et al., 2012). HLA receptors are essential for vaccine development, as they are responsible for presenting antigenic peptides to T-cells, thus activating both humoral and cellular immune responses necessary for robust immunity (Jia et al., 2022). To assess the interaction and binding affinity of a newly designed MEV, molecular docking studies were performed using the ClusPro server by using
The analysis were focused on TLRs (TLR2: 6NIG, TLR3: 2A0Z, TLR4: 3FXI, TLR8: 3W3M) and HLA (5WJL) (Kozakov et al., 2017). The results were further validated through the HADDOCK server (De Vries et al., 2010).
Normal mode analysis and dynamic simulation
The normal mode analysis (NMA) of the model MEV were simulated by iMODS online tool in December 2024 (Ali et al., 2024c). The examination focused on key parameters such as the covariance matrix, eigenvalues, backbone deformation diagram, elastic B-factor values, and the network model. These aspects collectively offer a thorough insight into the dynamic properties of the modeled vaccine–receptor complexes.
A 100 ns MD simulation was performed using GROMACS 5.3 (2022) to assess the stability and consistency of the predicted vaccine construct structure (Van Der Spoel et al., 2005). Additionally, a simulation of the template peptide, utilized for vaccine structure prediction, was passed out to associate trajectory data between the modeled and predicted structures. This comparison aids in understanding the similarities in their trajectories. The structure topology was generated using the all-atom Optimized Potentials for Liquid Simulations force field.
Immune simulation
The MEV immunogenic quality was assessed in a real-world context using the C-ImmSim program, an antigen-based model designed to predict immunological responses through an artificial algorithm (Castiglione and Bernaschi, 2004). A four-week interval between doses is typically recommended for most immunizations. In this study, three doses were administered at four-week intervals, aligning with real-world vaccination schedules. Each dose contained 1,000 antigens, ensuring an optimal immune response following the initial injection. These injections were simulated at 1,234 time-steps, each corresponding to a 10 h real-world interval. All other parameters were kept constant, while the number of simulation steps was increased to 1,000.
In silico cloning
Codon optimization through the JCAT platform enhanced protein expression in E. coli K12 (Grote et al., 2005). XhoI and NdeI restriction sites were incorporated at the N- and C-termini of the vaccine sequence, respectively. The SnapGene software was employed to analyze the modified nucleotide sequence of the vaccine construct and its alignment with the E. coli pET28a(+) expression vector during the cloning process (Ali et al., 2023c).
Results
Retrieval of sequence
The HMPV genotypes (A2a, A2b, and A2c) were retrieved from NCBI. For vaccine design, the top two antigenic sequences of glycoproteins were selected from each genotype.
Prediction of T-cell epitopes
T-cell epitopes, composed of short peptides, can activate immune responses by binding to specialized receptors on CTLs. These receptors recognize antigens presented on the surface of infected cells, forming a specific complex. Using the IEDB server, conserved sequences from three proteins were identified to design T-cell epitopes for MHC class I and II. Selected epitopes demonstrated high binding affinity, broad HLA compatibility, and favorable properties, including high antigenicity, nonallergenicity, nontoxicity, surface localization, and complete conservancy across HMPV genotypes (Supplementary Table S1). Three 9-mer CTL epitopes with conserved sequences were identified as potential vaccine targets due to their ability to engage CD8+ T-cells via HLA class I binding, promoting cellular immunity. Similarly, three 15-mer HTL epitopes with cytokine-inducing properties were chosen for their ability to bind HLA class II, stimulating both humoral and cellular responses through CD4+ T-cells (Supplementary Table S2).
B-cell epitopes
B-cell epitopes are pivotal in vaccine strategies designed to prevent specific diseases. For effective immune activation, these epitopes must exhibit antigenicity and be exposed on the surface. The epitopes derived from A2a, A2b, and A2c were selected through rigorous evaluation of their antigenic potential, allergenic risks, and toxicity profiles. Notably, all chosen epitopes are devoid of allergenic and toxic properties, highlighting their suitability as vaccine candidates (Supplementary Table S3). By interacting with B-cell receptors, these epitopes initiate humoral or cellular immune responses, which are fundamental to the development and effectiveness of vaccines.
Vaccine construction and physiochemical properties
The overlapping epitopes for CTL, HTL, and B-cells are detailed in Table 1. The red color shows the overlapped region. Two adjuvant sequences, HBHA protein and β-defensin, were incorporated into the construct designs, as visualized in Figure 2 and Supplementary Figure S1. The physicochemical attributes of the formulated vaccines are outlined in Table 2. Antigenicity assessments using VaxiJen 2.0 and ANTIGENpro indicate strong antigenic potential for both vaccines (V1 and V2). Results from ToxinPred2 and AllerTop 2.0 confirm their nontoxic and nonallergenic nature. Stability analyses reveal instability indices of 50.03 and 51.60, denoting robust stability, while aliphatic indices of 55.34 and 44.26 suggest favorable thermal stability. Furthermore, extended half-life predictions and excellent solubility confirmed by SOLpro underscore the overall viability of the vaccines (Magnan et al., 2009).

Represent the vaccine construct using HBHA adjuvants and overlapped epitopes.
The Highly Antigenic Overlapped Epitopes of Glycoproteins HMPV
HMPV, human metapneumovirus.
The Physiochemical Properties of Designed Multiepitope Vaccine
2D and 3D structure vaccine analysis
The immunological and physicochemical potential of the V1 and V2 vaccine constructs led to their prioritization for advanced analysis. Using PSIPRED and Swiss model tools, secondary and tertiary structures were predicted, followed by precise refinement to achieve energy-minimized configurations with optimal Rama-favored profiles. Quality assessments revealed that the refined V1 and V2 structures aligned with acceptable Z-score ranges and adhered to energy-based validation criteria (Supplementary Fig. S3 and Fig. S4). Further structural refinement was carried out via the DeepRefiner web server, highlighting remarkable binding energy and stability. ERRAT quality ratings ranged from 58.33% to 97.43%, while ProSA-web Z-scores between −2.77 and −3.59 indicated superior structural quality (Wiederstein and Sippl, 2007). Comprehensive evaluations using RAMPAGE, ERRAT, and ProSA-web confirmed the reliability and accuracy of the modeled vaccine structures (Figs. 3 and 4, Supplementary Fig. S4 and Fig. S5, and Table 3).

The 3D structure and its refinement of multiepitope vaccine (MEV) V1.
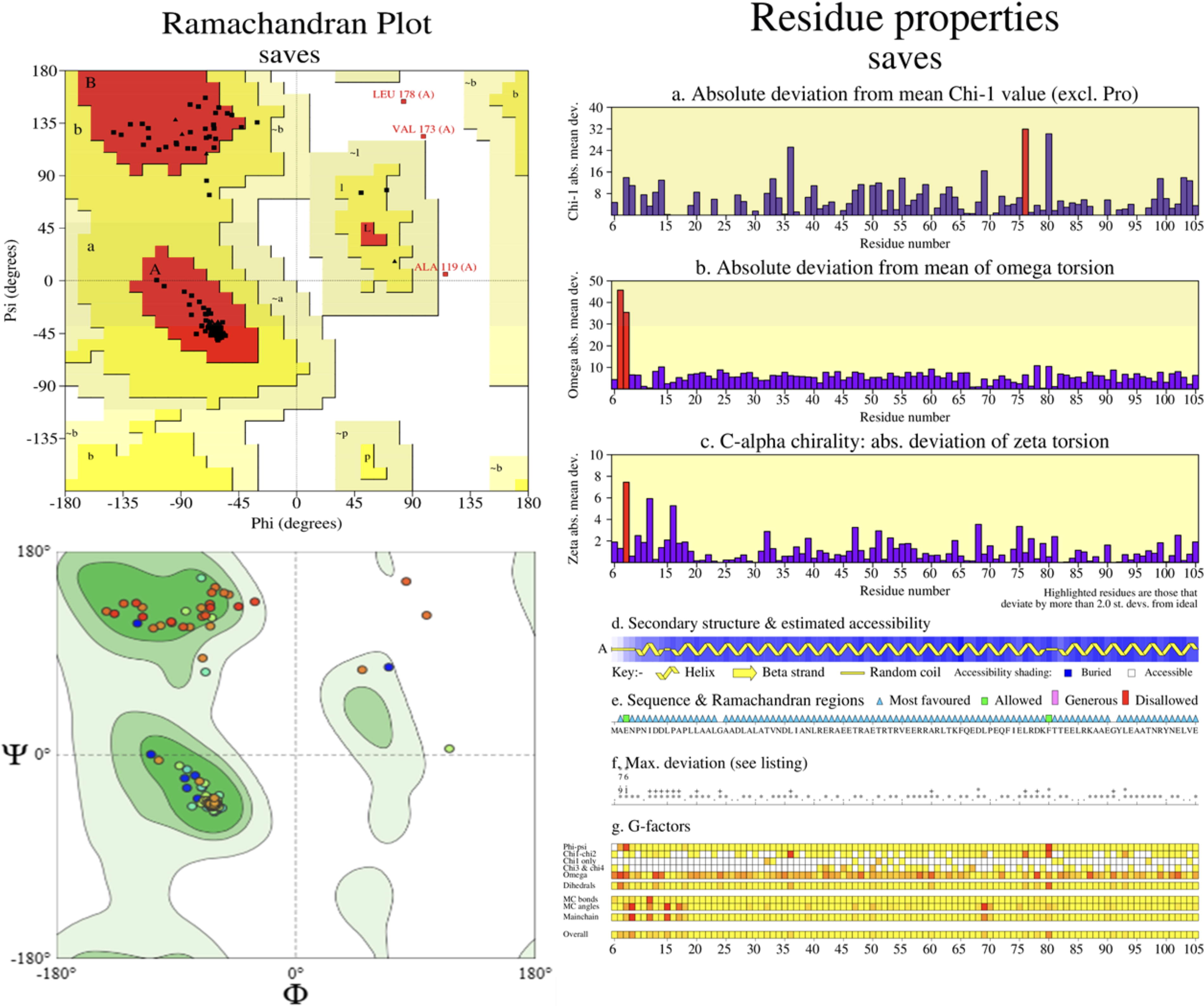
Present the Ramachandran plot and its favored region with residues demonstrate by graph of MEV V1.
The Protein PDB Structure Accuracy of MEV Models
MEV, multiepitope vaccine.
Molecular docking
The ClusPro 2.0 server was utilized to assess the binding affinity of vaccine constructs (V1 and V2) with receptor molecules, including HLA, TLR2, TLR3, TLR4, and TLR8. For each interaction, the server generated multiple candidate models, each associated with distinct binding energy value. The model exhibiting the lowest binding energy was selected for more optimization. Following this, the HADDOCK 2.4 platform was employed to refine the selected complex factors (Fig. 5).

HMPV-V1 molecular interaction with TLR8 and complex structure stability assessment.
The binding potential of the designed vaccine constructs was evaluated through docking simulations targeting HLA, TLR2, TLR3, TLR4, and TLR8 immune receptor molecules (Supplementary Fig. S6 and Fig. S7). Among the constructs, HMPV-V1 demonstrated the most favorable binding affinity with TLR8, registering an energy value of −15.8 kcal/mol (Tables 4 and 5). Additionally, Normal Mode Analysis (NMA) was used to assess the energy needed to deform the refined V1-TLR8 complex, which showed a high eigenvalue indicating structural rigidity and a low B-factor, suggesting reduced atomic fluctuations.
The Binding Score and Energies of HMPV-V1 and Receptors
HLA, human leukocyte antigen; TLR, toll-like receptor.
The Binding Score and Energies of HMPV-V2 and Receptors
Normal mode analysis
The MDs of the selected MEV docked with immune receptors were evaluated using NMA. This method examines the relationship between eigenvalues, representing vibrational frequencies, and B-factors, which indicate atomic or residue flexibility. Eigenvalues inversely correlate with flexibility, where lower values denote greater flexibility and higher values indicate increased rigidity. Similarly, B-factors, derived from X-ray crystallography or cryo-EM data, reflect atomic displacement, with higher values corresponding to flexible regions. Low-frequency modes (low eigenvalues) align with high B-factors, indicating flexibility, while high-frequency modes (high eigenvalues) correspond to rigidity. This inverse relationship validates computational molecular models against experimental data and aids in understanding structural dynamics, offering insights into vaccine–receptor interactions, stability, and functional modulation.
The study prioritized V1-TLR8 complexes based on minimal binding energy with immune receptors. The deformity graph identified main chain regions with structural hinges or linkers as peaks. The variance graph, linked to eigenvalues, displayed individual and cumulative variances for normal modes, while the covariance matrix illustrated atomic motion correlations, anticorrelations, and independence. Elastic network models highlighted atomic connectivity, with darker gray springs denoting higher stiffness. These analyses collectively demonstrated the significant stability of the V1 vaccine construct when complexed with TLR8, underscoring its potential efficacy and suitability for vaccine development (Fig. 6).
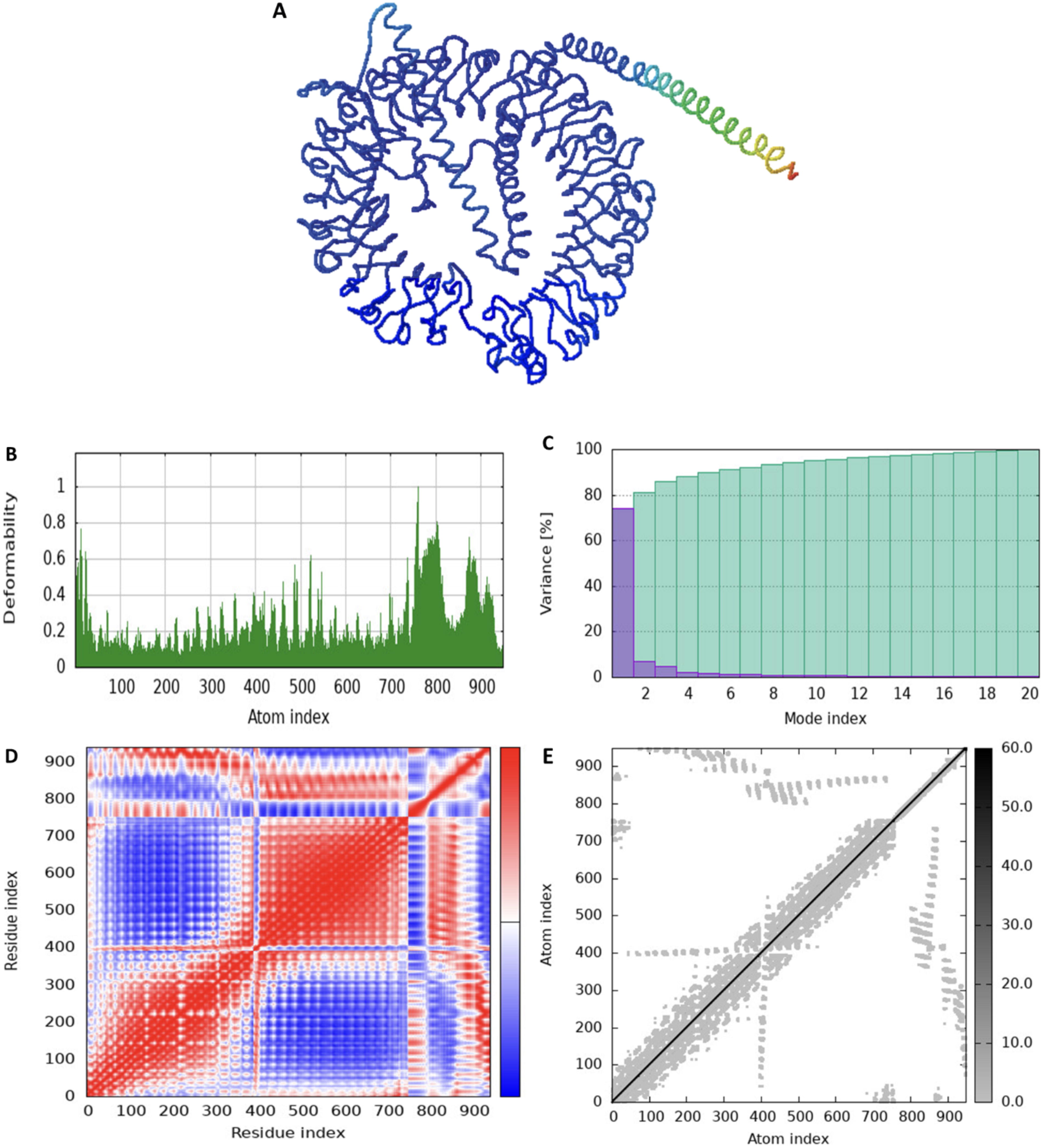
Normal mode analysis (NMA) results for V1-TLR8 complex were generated using the iMODS platform.
Molecular dynamic simulation
Molecular interactions between the model vaccine construct and receptor molecules were analyzed using Root Mean Square Deviation (RMSD), RMSF, and Radius of Gyration (Rg) metrics derived from MD simulations to evaluate the complex’s flexibility and stability. RMSD results demonstrated rapid stabilization, with structural deviations ranging from 0.1 to 0.4 nm following a 100 ns equilibration phase. Root Mean Square Fluctuation (RMSF) analysis revealed consistent rigidity in amino acid residues, signifying a stable configuration throughout the simulation. Thermodynamic assessments conducted using GROMACS provided insights into the system’s energetic stability and intermolecular interactions. The Rg evaluation further confirmed the compact and robust nature of the complex, showing minimal structural fluctuations. These findings underscore the dynamic stability and adaptability of the vaccine–receptor system, highlighting its potential efficacy in eliciting immune responses (Fig. 7).

Molecular dynamics (MD) simulations of the vaccine–receptor complex provided critical insights into their interactions.
Immune simulation
The evaluation of antibody levels offers valuable information regarding vaccine efficacy and immune responses to infections. Examining the dynamics of CTL and HTL provides clarity on the impact of vaccines on immune reactions. Results demonstrated a notable increase in immunoglobulin M (IgM) and IgG levels after 30 days of antigen stimulation, while a less pronounced rise in IgG2 was observed (Fig. 8A). Additionally, B-memory (y2) and B-isotype IgM levels were elevated (Fig. 8B). Furthermore, increases in TH memory (y2), TC, and TH cell populations were evident (Fig. 8C and D). The IFN-γ level peaked after 15 days of stimulation (Fig. 8E).

Immunization was simulated using C-ImmSim.
In silico cloning and codon optimization
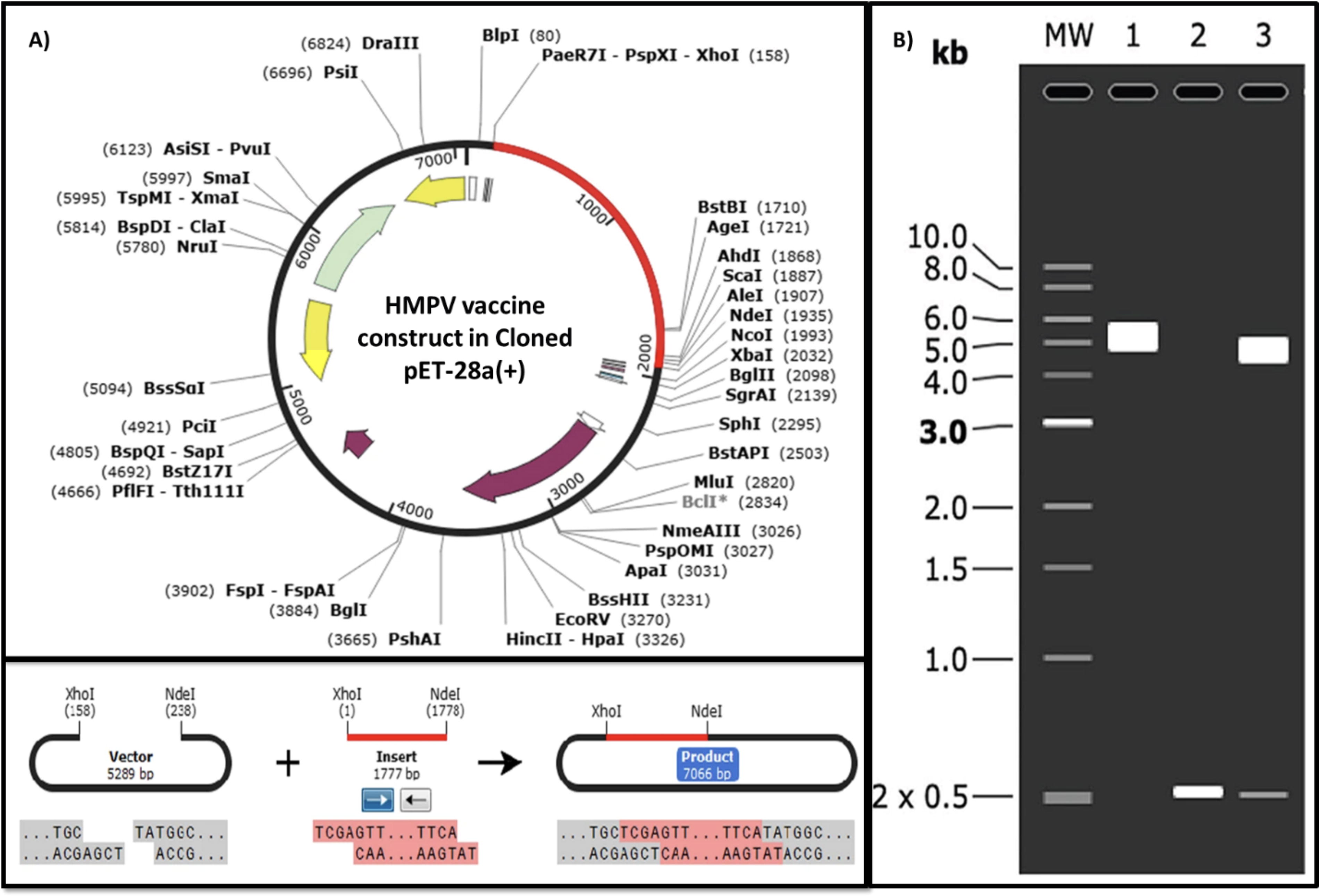
The Guanine-Cytosine (GC) content of the V1-TLR8 vaccine, optimized for genetic code, is 55.1525%, which falls within the optimal range. Both model vaccines demonstrated a Codon Adaptation Index of 1.0, indicating their potential for effective expression in the E. coli K12 strain. To enable the insertion of these constructs into the pET28(a)+ plasmid, restriction sites were incorporated at both the N- and C-termini (Fig. 9A). This precise genetic modification guarantees efficient cloning and expression of the selected vaccine model. Verification of the clones was performed using the SnapGene agarose gel simulation tool. Following digestion with SalI and KpnI enzymes, both the inserts (0.5 kb) and vectors (5.0 kb) were present as expected, with the insert size matching the calculated molecular weight of the candidate vaccine (Fig. 9B). Finally, the NCBI BLAST confirmed no sequence homology between the vaccine and host.
Discussion
HMPV is a major respiratory pathogen contributing to significant morbidity and mortality, particularly among young children, the elderly, and immunocompromised individuals (Bhasin et al., 2024). It is categorized into two primary genotypes, A and B, which are further divided into sublineages, including A2a, A2b, and A2c. These sublineages exhibit notable genetic variation and distinct epidemiological behaviors, with A2a and A2b linked to moderate to severe respiratory infections, while A2c is associated with heightened virulence and a potential for broader outbreaks (ALi et al., 2023; Mizuta et al., 2010). Despite its clinical significance, no approved vaccine is available for HMPV, highlighting the critical need for effective preventive measures. This study aims to develop a MEV targeting conserved, immunodominant regions of HMPV, utilizing advanced immunoinformatics techniques to provide comprehensive protection against the virus and its diverse sublineages (Ma et al., 2024).
The present study began by predicting immunogenic epitopes from high-ranking vaccine candidate proteins of HMPV (A2a, A2b, and A2c), focusing on glycoproteins critical for the virus lifecycle. Targeting these proteins offers a promising approach for preventing HMPV infection. Two glycoproteins from each genotype were chosen, resulting in six proteins for MEV design. Eighteen highly antigenic and overlapping epitopes were selected from these proteins to construct two vaccine models, each with distinct adjuvant sequences. T-cell epitopes that overlapped with B-cell epitopes and had the potential to induce IFN-γ were prioritized. These epitopes were predicted to stimulate T-cell, B-cell, and IFN-γ mediated immune responses (Kak et al., 2018). Toxicity and allergenicity assessments ensured the safety of the selected epitopes. The epitopes were then integrated with appropriate linkers and adjuvants, systematically evaluated to optimize their combinations for effective vaccine models. The selection of adjuvants was based on their immune-enhancing properties, safety, and mechanism of action. Linker sequences, which connect various antigenic or adjuvant components, were chosen for flexibility, stability, and immunogenicity. The chimeric vaccine constructs incorporated top epitopes and specific linkers such as EAAAK, GGGS, HEYGAEALERAG, and PADRE sequences, including “AKFVAAWTLKAAA.” EAAAK and PADRE linkers were selected for their established efficacy in enhancing immunogenic potential (Ali et al., 2025). The EAAAK linker improves flexibility and antigen presentation, while the PADRE linker ensures broad immune activation by interacting with diverse Human Leukocyte Antigen – DR isotype (a class II MHC molecule) (HLA-DR) molecules, crucial for targeting varied populations (Ali et al., 2024). These linkers have demonstrated effectiveness in previous studies and were selected to enhance expression, solubility, and proper folding of the vaccine’s 3D structures. The combination of epitopes and linkers facilitated the creation of multiple vaccine constructs, optimizing expression, bioactivity, and robust immune responses for vaccine development (Ali et al., 2024).
The vaccine design incorporates a range of B-cell, CTL, and HTL epitopes, along with linkers and β-defensive adjuvants to enhance immunogenicity. The top-ranking vaccine candidates exhibit high antigenicity and are free from allergenic and toxic effects. The leading candidate stands out for its molecular stability, basicity, and hydrophobicity, which are key to promoting a strong immune response. Following refinement, the 3D structures of the proposed HMPV vaccine models show significant improvements in stability, as verified by Ramachandran plot analysis (Zhuang et al., 2024a). Binding affinity assessments of the vaccine models against HLA, TLR2, TLR3, TLR4, and TLR8 demonstrate favorable interactions with these immune receptors. Notably, TLR8 is capable of recognizing single-stranded RNA from viruses (Atukpa et al., 2024; Aziz et al., 2024). These findings suggest that the vaccine molecules may effectively suppress HMPV infection by triggering the production of type-1 interferon, inflammatory cytokines, and chemokines. Previous studies have highlighted the crucial role of HLA molecules in enhancing both humoral and cellular immune responses to infections.
Molecular docking studies with TLR and HLA receptors identified significant interactions, particularly with TLR4 and HLA, supporting the potential of the HMPV V1 lead vaccine construct in eliciting strong immune responses (Zhuang et al., 2024). The iMODS (NMA) server proves valuable for analyzing the intrinsic dynamics of proteins, offering a detailed assessment through parameters such as eigenvalues, covariance, B-factors, and deformability, which describe protein motion and stability. Eigenvalues, indicating primary chain deformability, correlate with the energy required for conformational changes, with smaller values suggesting increased susceptibility to bending (Ali et al., 2023). Covariance analysis highlights the coordinated movements between different protein regions. Higher B-factors reflect enhanced flexibility, a critical factor in assessing the viability of protein complexes for biomedical applications, including vaccine design (Manzoor et al., 2023). The peptide sequence HMPV V1 showed an eigenvalue of 2.536854e-06, signifying increased flexibility in the vaccine model. MDs simulations confirmed the favorable interactions and stability of the vaccine model, particularly with TLR8 immune receptor. Immune simulations suggested promising immune responses for the HMPV V1 vaccine construct. The physicochemical properties of these constructs remained stable after expression, ensuring high solubility and stability (Aiman et al., 2023; Awais et al., 2023). Recombinant peptide overexpression in E. coli is essential for functional and biochemical characterization, while serological testing is necessary for confirming vaccine efficacy. The E. coli system is optimal for recombinant peptide cloning and development. These results provide a critical framework for the design of next-generation vaccines targeting the HMPV virus.
Limitations
This study provides a computational framework for MEV design, certain limitations must be acknowledged. The predicted vaccine candidates require in vitro and in vivo validation to confirm immunogenicity and stability, followed by clinical trials to assess real-world efficacy, safety, and immune response in human subjects. Additionally, the selection of adjuvants and delivery mechanisms needs further optimization through experimental evaluation to enhance vaccine formulation. Future research should address these limitations to advance the vaccine candidate toward clinical application.
Conclusions
This study presents a comprehensive framework for designing a next-generation MEV against HMPV, addressing the urgent need for effective prophylactic interventions. By targeting conserved and immunodominant epitopes of glycoproteins from major HMPV sublineages (A2a, A2b, and A2c), the proposed vaccine constructs demonstrate high antigenicity, molecular stability, and safety. Rigorous immunoinformatics-driven analyses, including epitope prediction, molecular docking, and dynamics simulations, confirm the vaccine’s robust potential to elicit both humoral and cellular immune responses through interactions with key immune receptors such as TLR2, TLR3, TLR4, TLR8, and HLA molecules.
The incorporation of optimized linkers and adjuvants further enhances the immunogenic profile, stability, and expression potential of the vaccine models. Immune simulations indicate promising responses, supporting the constructs’ ability to trigger a protective immune defense. The constructs’ solubility and stability in an E. coli expression system suggest feasibility for large-scale production and subsequent experimental validation. These findings provide a solid foundation for advancing vaccine development against HMPV, offering a promising strategy to mitigate the global burden of HMPV-associated respiratory infections. Future experimental studies and clinical trials are imperative to translate these computationally designed constructs into effective therapeutic solutions.
Footnotes
Acknowledgments
The authors gratefully acknowledge the Abdul Wali Khan University Mardan, KPK, Pakistan.
Authors’ Contributions
E.M.K. and S.Y.A.: Conceptualized the study, conducted the research, and cowrote the article. A.A., M.A.A.M., and S.L.A.: Provided critical insights, supervised the research, contributed to the article’s intellectual content and participated in research, data analysis, and preparation of the article. All the authors reviewed and approved the final article.
Data Analysis Sources
Genbank, NCBI (https://www.ncbi.nlm.nih.gov/genbank/), virus accession ID: 683172; IEDB web server (https://www.iedb.org/); ABCpred web server (http://webs.iiitd.edu.in/raghava/abcpred/); VaxiJen v3.0 web server (https://www.ddg-pharmfac.net/vaxijen3/); AllergenPro web server (http://nabic.rda.go.kr/allergen/); AllerTop v.2.0 web server (https://www.ddg-pharmfac.net/AllerTOP/); ToxinPred v3.0 web server (https://webs.iiitd.edu.in/raghava/toxinpred/); ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php); Protein Data Bank (PDB) sum web server (https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/); PSIPRED web server (http://bioinf.cs.ucl.ac.uk/psipred/); HawkDock web server (http://cadd.zju.edu.cn/hawkdock/); Hdock web server (http://hdock.phys.hust.edu.cn/); iMODS web server (http://imods.chaconlab.org/); C-ImmSim server (https://kraken.iac.rm.cnr.it/C-IMMSIM/?page=0); and JCat web server (![]() ).
).
Data Availability
Data used in this study are available in the article and its supplementary files.
Author Disclosure Statement
The authors declare no conflicts of interest related to this work.
Funding Information
No funding received for this research.
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.