
Abstract
Objective:
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease that affects 63 in every 100,000 Americans. Its etiology remains unknown, although inflammatory pathways appear to be important. Given the dynamic environment of the lung, we examined the significance of mechanotransduction on both inflammatory and fibrotic signaling during IPF.
Innovation:
Mechanotransduction pathways have not been thoroughly examined in the context of lung disease, and pharmacologic approaches for IPF do not currently target these pathways. The interplay between mechanical strain and inflammation in pulmonary fibrosis remains incompletely understood.
Approach:
In this study, we used conditional KO mice to block mechanotransduction by knocking out Focal Adhesion Kinase (FAK) expression in fibroblasts, followed by induction of pulmonary fibrosis using bleomycin. We examined both normal human and human IPF fibroblasts and used immunohistochemistry, quantitative real-time polymerase chain reaction, and Western Blot to evaluate the effects of FAK inhibitor (FAK-I) on modulating fibrotic and inflammatory genes.
Results:
Our data indicate that the deletion of FAK in mice reduces expression of fibrotic and inflammatory genes in lungs. Similarly, mechanical straining in normal human lung fibroblasts activates inflammatory and fibrotic pathways. The FAK inhibition decreases these signals but has a less effect on IPF fibroblasts as compared with normal human fibroblasts.
Conclusion:
Administering FAK-I at early stages of fibrosis may attenuate the FAK-mediated fibrotic response pathway in IPF, potentially mediating disease progression.

INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a significant health care burden, costing the U.S. health care system ∼$2 billion a year. 1 The IPF incidence in the United States is estimated to affect 63 individuals per every 100,000 in the population. 2 The IPF is associated with high morbidity and mortality, with a mean survival rate of 2–5 years from the time of diagnosis. 3 Estimated mortality rates are 64.3 deaths per million in men and 58.4 deaths per million in women. 4 Currently, there are a few effective therapeutic or preventive pharmacological agents. 2
Lung fibrosis is a progressive disease leading to organ failure and death. 5 Fibrotic processes are believed to be the result of an exuberant response to injury that stimulates the proliferation, migration, and differentiation of mesenchymal cells that become fibrogenic myofibroblasts. 5 Myofibroblast-rich tissues then develop into non-functional scar lesions with stiffer mechanical properties. 5 Lung myofibroblasts experience a dynamic physical environment, and therefore understanding the fundamental mechanisms by which these cells are generated and activated is essential for the development of new therapies.
Although the mechanisms of the pathophysiology of fibrosis continue to be studied, recent findings suggest that Focal Adhesion Kinase (FAK) plays a key role in regulating fibrotic signaling and mechanotransduction. 6 –8 The FAK is activated integrin-mediated adhesion and is at the intersection of integrin and growth factor receptor signaling cascades. 9 The FAK is crucial in cell proliferation, differentiation, migration, and survival. 9 The FAK activation increases the expression of genes related to mechanotransduction and extracellular matrix (ECM) deposition, leading to increased prevalence of fibrogenic pathways that are important for scar formation after injury. 10 The FAK drives the progression of liver fibrosis by modulating the proliferation, activation, and apoptosis of hepatic stellate cells. 11 The FAK has also been attributed as a key player in modulating lung fibroblasts. 12 Specifically, fibrotic lung fibroblasts migrate faster than normal human lung fibroblasts, 13 a property that could explain the development of progressive forms of IPF. Due to FAK's critical role in an array of functions, targeting the FAK pathway may be a promising therapeutic target for treating fibrotic diseases such as IPF.
In this study, we characterized the effects of mechanical strain and the role of FAK in modulating the interconnected network of pro-inflammatory and pro-fibrotic genes in the lung. We used a combination of in vivo and in vitro approaches by utilizing fibroblast-specific FAK knock out (FAK-KO) in murine lung fibrosis and pharmacologic FAK inhibitors in human normal and IPF fibroblasts. We utilized an established in vitro culture system to manipulate the mechanical strain applied to normal and IPF human fibroblasts and then examined the effect of chemical FAK inhibitor (FAK-I). Transcriptional profiles were analyzed by using quantitative real-time polymerase chain reaction (qRT-PCR), Western Blot, histology, and pathway analysis (Fig. 1). Our findings suggest that mechanical strain may be an important mediator of pro-inflammatory and pro-fibrotic transcriptional pathways. Further studies of how mechanotransduction affects human IPF fibroblasts may lead to novel therapeutic targets for the treatment of IPF.
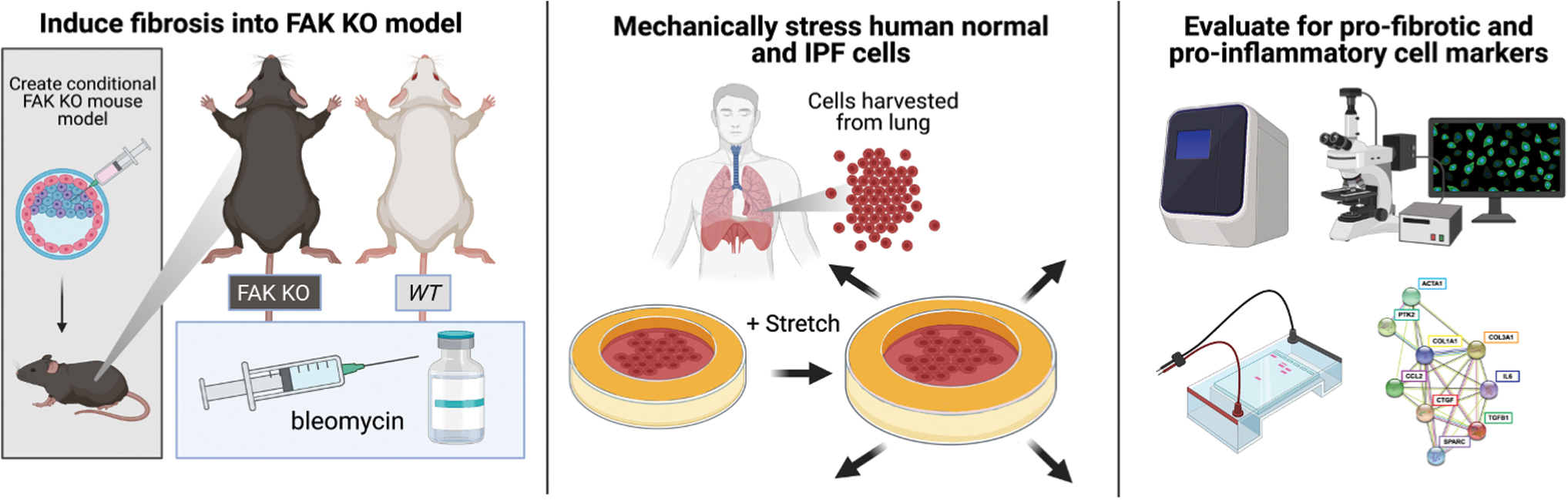
Summary graphical overview illustrating the role of FAK and interconnected mediators in both human and mouse models using robust quantification analytics. To further understand the role FAK has in modulating the fibrotic response in induced pulmonary fibrosis (IPF), a multi-pronged approach was taken in both human and mouse models. FAK-KO mice were subjected to a bleomycin-induced pulmonary fibrosis model. Human cells were subjected to mechanical strain and were evaluated for pro-fibrotic and pro-inflammatory markers by using a variety of robust analytical techniques. FAK-KO, Focal Adhesion Kinase-knock out; IPF, idiopathic pulmonary fibrosis. Color images are available online.
INNOVATION
Patients suffering with IPF do not have access to robust, clinically effective pharmacotherapies that can alleviate and reverse effects of fibrosis. One way to tackle this ongoing clinical challenge is to further elucidate the role of key regulators that may be important in its pathophysiology, such as mechanotransduction pathways. Although acute models of pulmonary fibrosis do not necessarily predict clinical outcome of a chronic disease such as IPF, recent studies have identified small-molecule inhibitors for mechanotransduction pathways 7,14 and these therapeutics may be well suited as new treatments to mitigate fibrosis and IPF.
CLINICAL PROBLEM ADDRESSED
The effects of mechanical strain on cellular differentiation and transcription affect the development of fibrosis in IPF. Understanding how to mitigate mechanotransduction may help us develop therapies to target FAK-mediated fibrotic and inflammatory response pathways.
MATERIALS AND METHODS
Animals
All 8–10-week-old mice were housed in the Stanford University Veterinary Service Center in accordance with NIH and institution-approved animal care guidelines. All procedures were approved by the Stanford Administrative Panel on Laboratory Animal Care. A conditional, fibroblast-specific FAK-KO mouse model was used, as previously described. 7 Briefly, we created conditional KO mice with fibroblast-restricted FAK deletion by crossing heterozygous fibroblast-specific procollagen-α2(I)–Cre recombinase mice to homozygous floxed FAK mice (B6.129-Ptk2tm1Lfr/Mmcd, purchased from the UC Davis Mutant Mouse Regional Resource Center). For bleomycin and control (phosphate buffered saline [PBS]) experiments, homozygous floxed FAK mice (Cre negative) were used. We backcrossed mice at least eight generations on a C57BL/6 background, identified progeny, and administered tamoxifen injection in all mice through daily intraperitoneal injections for 5 days 1 week before experiments by using published methods. 15
Human cells
Human normal and IPF cells were kindly provided by the Stanford Center for Advanced Lung Disease as a gift. A total of eight individual samples were used (four normal and four IPF). The IPF cells were isolated and harvested according to an established protocol in the literature. 16 Cells were cultured in Fibroblast Growth Medium (cat. no. 116–500; Sigma Aldrich, St. Louis, MO) in six-well plates, and cultures were expanded for several passages by using a standard culture method. For in vitro experiments, only cells between passage numbers 3 and 5 were used to generate data. Fibroblasts were characterized as expressing basal level of alpha-smooth muscle actin (α-SMA) at their quiescent state in culture, and this expression was detected by using immunofluorescence (data not shown). All in vitro cell experiments were conducted in triplicates.
Bleomycin-injected lung fibrosis mouse model
Mice were divided randomly into the following three groups: (A) saline-only (n = 5, control group, Control), (B) bleomycin 1 mg/kg (n = 5, bleomycin-treated group, Bleo), and (C) FAK-KO bleomycin 1 mg/kg (n = 5, bleomycin-treated group with fibroblast-restricted FAK knock out, FAK-KO+Bleo). The experiment was repeated with n = 8 mice in each group for quantitative polymerase chain reaction (qPCR) analysis. To induce pulmonary fibrosis, bleomycin hydrochloride (Bleomycin HCl, Nippon Kayaku, Tokyo, Japan) was dissolved in sterilized 0.9% saline and administered intratracheally as a single dose of 1 mg/kg in 30 μL solution per animal. The Control group was treated with 30 μL of sterile 0.9% saline instead of bleomycin on day 0. To investigate the effects of FAK on pulmonary fibrosis development in vivo, we used a widely used model for lung fibrosis, intratracheal injection of a single dose of bleomycin to induce lung injury. 17 –19 Both lung lobes of Control, Bleo, and FAK-KO+Bleo mice were harvested on D21 for histology and independent qPCR analysis.
Histology and staining
On Day 21, mouse lungs were harvested and fixed in paraformaldehyde. They were subsequently dehydrated with a graded alcohol series, embedded in paraffin, and sectioned into 8-μm-thick sections. 20 For all immunochemical analyses, samples were deparaffinized and subsequently stained. Masson's trichrome staining (Sigma-Aldrich) was used to evaluate collagen networks (n = 5 per group). α-SMA immunofluorescent staining of cross-sectional mouse lung tissue slices was performed, n = 5 for all groups (A5228; Sigma-Aldrich).
Immunofluorescent staining of human normal and IPF fibroblasts was performed (n = 4 all groups) to quantify pro-inflammatory/pro-fibrotic expression. For each condition, three high power fields at 20 × were examined for each sample. Briefly, slides underwent antigen retrieval by using 0.01 M sodium citrate buffer in PBS solution, followed by blocking for 1 h with 5% goat serum in PBS. Slides were incubated with anti-Ki67 (SAB5700770; Sigma-Aldrich), α-SMA (A5228; Sigma-Aldrich), and Collagen I (SAB4500362; Sigma-Aldrich) antibodies overnight followed by incubation with a secondary antibody. Nuclei were stained with Flouroshield with DAPI (F6057; Sigma-Aldrich). ImageJ was used to quantify staining intensity. All measurements were performed by an individual blinded to the treatment group.
Assessing scar formation and collagen deposition using FracLac
The complexity and heterogeneity in Masson's Trichrome tissue samples from Control, Bleo, and FAK-KO+Bleo mice were measured by using the ImageJ plug-in FracLac. 21 Local fractal dimensions (FD) and lacunarity (L) values were calculated by using the subsample box counting scan (50 grid default sampling size, minimum pixel density threshold = 0, rectangle subscan). The FD measures the density of collagen networks. A higher FD has a denser and scar-like fiber arrangement. L measures the amount of randomness or heterogeneity in a sample. A low L value implies less heterogenous collagen fiber orientation.
Ashcroft scale
Lung sections were assessed by a system of modified grades, as previously described. 22 Briefly, a numerical scale for determining the degree of fibrosis in lung tissue was used that evaluated both the alveolar septa and lung tissue structure. Grading was scored on a scale from 0 to 8, with 0 representing no fibrosis and 8 representing significant fibrosis.
Gene expression analysis with qPCR
D21 mouse lung tissue samples for Bleo and Bleo+FAK-KO were snap frozen on dry ice and stored at −80°C. Gene Expression Assays (Applied Biosystems, Foster City, CA) were used for in vitro transcriptional analyses (n = 8 for all gene expression assay groups). Assays were performed for Acta2 (Mm00725412_s1), Col1a1 (Mm00801666_g1), Ptk2 (Mm00433209_m1), and Tgfb1 (Mm01178820_m1). Assays were repeated on human lung fibroblasts isolated at the terminal timepoint of 2 h after mechanical strain to evaluate pro-fibrotic and pro-inflammatory markers CCN2 (Hs00170014_m1), ACTA2 (Hs00426835_g1), TGFB1 (Hs00998133_m1), CCL2 (Hs00234140_m1), and IL6 (Hs00174131_m1). Treatment groups included no strain (NS), strain alone (S), and strain+FAK-I (S+FAK-I), n = 8 for all qRT-PCR samples.
Western blot
Both mouse and human lung cells underwent the same extraction protocol. Mouse fibroblasts were isolated from D21 lung tissue. Human fibroblasts were isolated 2 h 23 after multiaxial mechanical strain. Both fibroblast types were homogenized in 1 mL of RIPA lysis buffer (Sigma-Aldrich) and centrifuged. The aqueous layer was collected, and protein concentration was determined by using the BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL). Equal amounts of protein (20 μg) were strained into each lane of a gel. The separated proteins were transferred to a nitrocellulose membrane, and immunostaining was performed for p-FAK (44–624G), FAK (39-6500), α-SMA (MA5-11547), and α-tubulin (A11126). Band densities were quantified with ImageJ software (National Institutes of Health, Bethesda, MD).
Mechanical strain experiment
A customized stretching device was developed in our laboratory to apply periodical mechanical strain to an elastomeric membrane that served as a cell culture substrate. Human normal and IPF fibroblasts were plated on a 20 × 20 mm poly(dimethylsiloxane) (PDMS) (Sylgard, Corning, Midland, MI) membrane with an elastic modulus of ∼1 MPa. Previous work has demonstrated the suitability of PDMS dishes. 24 –26 Before the experiments, the stretching chambers were sterilized with ethanol, washed several times with PBS, and incubated overnight (12–14 h) with a 5 μg/mL Fibronectin solution (Sigma-Aldrich). Cells were plated at a density of 50 cells/mm2. On 80% confluency, 10% multiaxial strain was applied on human lung fibroblasts for a period of 2 h. Cells were subsequently isolated for further analyses. Markers in the center region of the stretching chambers were used to find the same position on the membrane in all experiments. Treatment groups included no strain (NS), strain alone (S), and S+FAK-I. FAK-I (PF-573228) was administered at a concentration of 10 μM for 1 h treatment in vitro.
Statistical analysis and laboratory notebook
All values are expressed as mean ± standard error of the mean. Statistical significance was determined by using a two-tailed unpaired t-test for experiments involving only two conditions. For experiments with three or more conditions, pairwise t-tests were employed, and p-values were adjusted by using Bonferroni correction for multiple hypothesis testing. A p-value <0.05 was considered statistically significant. An electronic laboratory notebook was not used.
RESULTS
FAK-KO mice are resistant to developing acute pulmonary fibrosis caused by intratracheal bleomycin injection
To elucidate the role of FAK in pulmonary fibrosis, we implemented a widely used in vivo experimental model of lung fibrosis by using an intratracheal injection of a single dose of bleomycin 7 (Fig. 2A). Masson's trichrome staining confirmed that bleomycin-treated lung tissue isolated on D21 exhibited an increased amount of densely packed collagen networks, noted by thickened alveolar septa and excessive deposition of fibrous materials, indicative of detrimental tissue remodeling (Fig. 2B). In the FAK-KO group, we observed a considerable reduction in the amount of collagen deposition and the density of alveolar collagen, demonstrating that FAK signaling may play a significant role in the development of pulmonary fibrosis.

FAK-KO mice are resistant to developing acute pulmonary fibrosis caused by an intratracheal bleomycin injection.
To quantitatively measure the extent and severity of fibrosis, the lungs were analyzed by using Modified Ashcroft Scores (Fig. 2C). This scoring system is used clinically and is a quantification method for analyzing bleomycin-induced fibrotic lung tissue in mice, 22 with higher numbers representing increased fibrosis. Fibrotic scores of FAK-KO+Bleo mice were significantly lower (2.80 ± 0.25, ***p = 0.001) than in Bleo only (5.99 ± 0.15, ***p = 0.001), although still higher than control lungs (0.50 ± 0.10, ***p = 0.001). These data demonstrate that fibrotic pathological features as defined by a clinically relevant scoring system were attenuated in FAK-KO mice, returning alveolar tissue to a less fibrotic state.
We further quantified the bleomycin-induced structural changes of lung tissue by calculating the fractional dimension and lacunarity of the fibrous tissue (Fig. 2D, E). FAK-KO+Bleo tissue demonstrated a lower fractional dimension (1.64 ± 0.005), as compared with Bleo alone (1.67 ± 0.009, **p = 0.01), both of which were higher than normal lungs (1.56 ± 0.009, ***p = 0.001) (Fig. 2D). An increase in fractional dimension indicates severe changes to the normal mesh architecture of the alveolar septa, with a denser and scar-like alveolar collagen arrangement. FAK-KO significantly attenuated this fibrotic progression within the alveolar cells. In addition, the lacunarity observed in FAK-KO+Bleo tissue (0.27 ± 0.003) was higher than that observed in Bleo alone (0.25 ± 0.003, ***p = 0.001) but again lower than in normal lungs (0.29 ± 0.004, ***p = 0.001) (Fig. 2E). Further, an increased collagen intensity was observed in Bleo alone (597.25 ± 59.21), as compared with normal lungs (239.06 ± 42.36). A decrease was observed in the FAK-KO+Bleo tissue (342.83 ± 63.81) as compared with Bleo alone, returning to a value like that observed in control (control vs. FAK-KO+Bleo, no significance, p > 0.05) (Fig. 2F). This demonstrates that fibrous collagen deposition is greatest and most homogeneous in the Bleo-only group. Randomness in the collagen fiber arrangement increased in the FAK-KO+Bleo group, representing a reversal of the detrimental effects of bleomycin-induced fibrosis.
Fibrotic marker expression was attenuated in FAK-KO mice treated with bleomycin
We subsequently examined the changes in fibrogenic gene expression in both the Bleo and FAK-KO+Bleo groups. We found that the expression of Col1a1 (0.74 ± 0.01) and Tgfb1 (0.68 ± 0.01), two known genes driving fibrosis and inflammation, 27 was significantly downregulated in FAK-KO+Bleo mice (1.00 ± 0.01, ***p = 0.001; 1.003 ± 0.039, ***p = 0.001, respectively) (Fig. 3A). Tgfb1 has been previously shown to play a pivotal role in ECM remodeling in the lung via activation of resident lung fibroblasts. 28

Fibrotic marker expression was attenuated in FAK-KO mice treated with bleomycin.
A significant and expected decrease in Ptk2, which codes for FAK, was observed in the FAK-KO+Bleo (0.188 ± 0.024, ***p = 0.001) as compared with Bleo alone (1.013 ± 0.0719) (Fig. 3A). This was further corroborated at the protein level by using Western Blot analysis of protein isolated from our murine lung tissue samples (Fig. 3B). Acta2, which encodes for α-SMA and is a primary marker of myofibroblasts, was upregulated with bleomycin treatment (1.44 ± 0.16), but it was significantly decreased in the absence of FAK (0.81 ± 0.022, *p = 0.05) (Fig. 3A). Myofibroblasts may be primary effector cells in lung fibrosis, actively synthesizing fibrogenic proteins in response to lung injury. 29 –31 This was confirmed at the protein level, with immunofluorescent staining demonstrating a significant reduction in α-SMA myofibroblasts in the FAK-KO+Bleo group (12.691 ± 1.495, **p = 0.01), as compared with Bleo alone (29.537 ± 2.798), both of which were increased in comparison to control lung tissue (5.436 ± 0.427, **p = 0.01) (Fig. 3C, D). These findings suggest that the beneficial effects of FAK inhibition may be mediated in part by decreasing the number of myofibroblasts.
Human IPF cells display increased expression of profibrotic/inflammatory markers
We subsequently sought to evaluate the difference between human normal and IPF lung fibroblasts in vitro (Fig. 4A–E). We obtained both lung fibroblasts from patients undergoing lung or heart–lung transplant procedure and isolated cells from normal and IPF lungs. These normal and IPF human lung fibroblasts were cultured for 5 days, fixed, and finally stained (Fig. 4B). We observed that compared with the normal lung fibroblasts, proliferation was significantly increased in IPF cells (3.34 ± 0.922 vs. 7.45 ± 1.11, *p < 0.05) as quantified by Ki67 staining (Fig. 4C). We also observed an overall increase in the relative intensity of Collagen I and α-SMA signal in IPF cells, indicating an increase of myofibroblast differentiation and ECM deposition (99.57 ± 8.29, ***p < 0.001 vs. 36.69 ± 5.65; 13.64 ± 1.91, *p < 0.05 vs. 6.66 ± 0.89, respectively) (Fig. 4D, E). These findings confirm previous studies demonstrating an increased amount of myofibroblasts and fibrosis in IPF tissues. 32,33
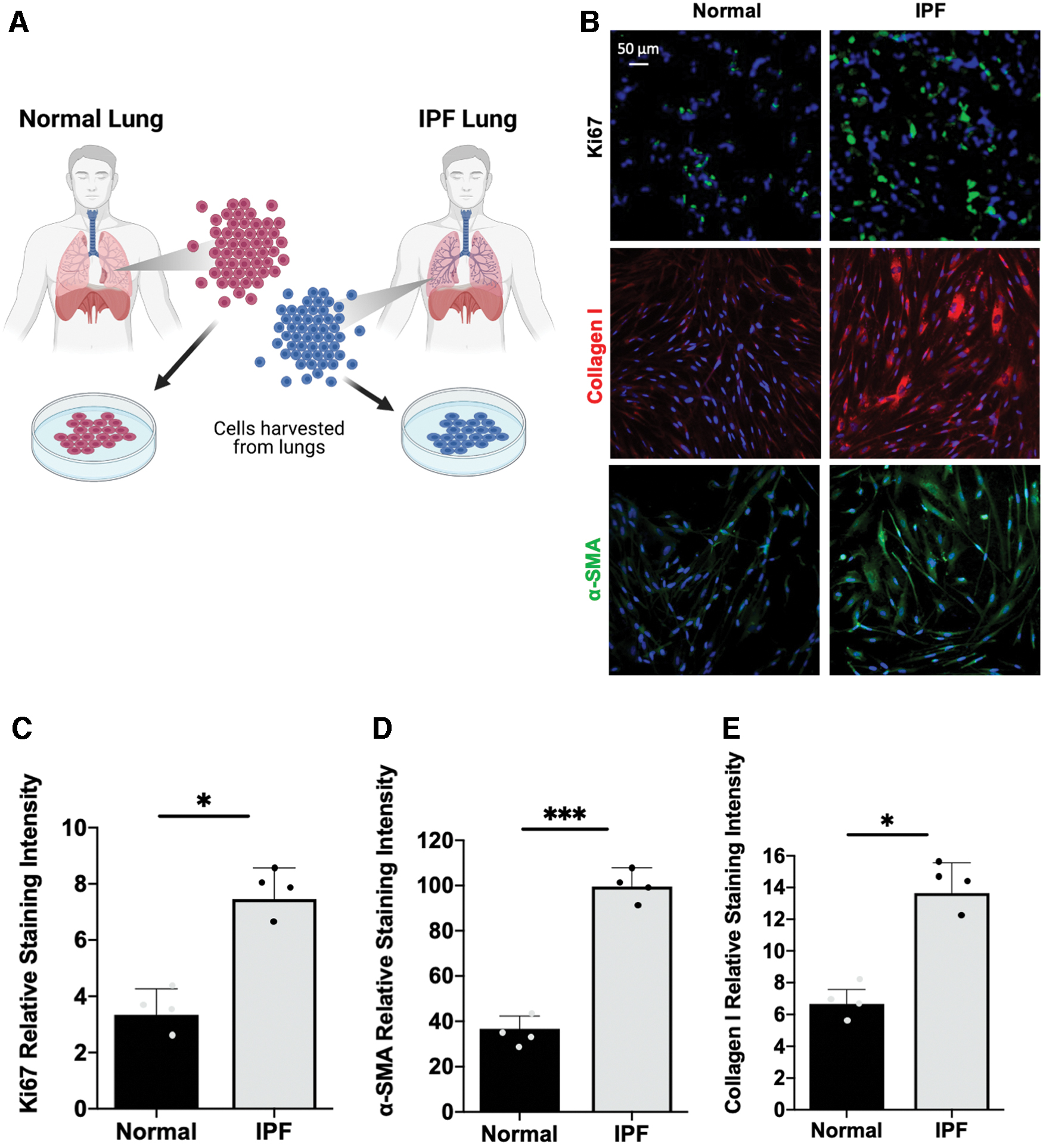
Human IPF cells display increased expression of pro-fibrotic/inflammatory markers.
FAK inhibitor treatment inhibits mechanical strain-induced increase in pro-fibrotic/inflammatory markers in human lung cells
To investigate mechanical signaling and the role of FAK in fibrosis, we utilized a well-described multiaxial mechanical strain system to quantify the activation of pro-fibrotic and pro-inflammatory cell markers in human normal and IPF lung cells 26 (Fig. 5A). In normal human fibroblasts, after 2 h, we observed an increase in the amount of p-FAK and α-SMA protein expression, as would be expected (Fig. 5B). Gene expression from cells isolated at the terminal timepoint of 2 h was quantified to catalog the differences in pro-fibrotic and pro-inflammatory gene expression (Fig. 5C–F).

FAK inhibitor treatment inhibits mechanical strain-induced increase in pro-fibrotic/inflammatory markers in human lung cells.
In normal human lung cells, when mechanical strain alone (S) was introduced, there was an upregulation of pro-fibrotic genes such as CCN2, ACTA2, and TGFB1 [CCN2 (1.77 ± 0.02, *p < 0.05 vs. 1.34 ± 0.02), ACTA2 (1.21 ± 0.04, *p < 0.05 vs. 1.11 ± 0.02) and TGFB1 (1.31 ± 0.02, *p < 0.05 vs. 0.98 ± 0.02)]. When mechanical strain (S) was introduced in the presence of FAK inhibition (S+FAK-I), the overall expression of these pro-fibrotic markers was substantially decreased [CCN2 (0.64 ± 0.01, ▲p < 0.05 vs. 1.77 ± 0.02), ACTA2 (0.94 ± 0.02, ▲p < 0.05 vs. 1.21 ± 0.04), and TGFB1 (0.84 ± 0.04, ▲p < 0.05 vs. 1.31 ± 0.02)] (Fig. 5C).
Strain also increased the expression of pro-inflammatory genes CCL2 and IL6 in normal human lung fibroblasts [CCL2 (0.42 ± 0.01, *p < 0.05 vs. 0.35 ± 0.02), IL6 (0.28 ± 0.006, *p < 0.05 vs. 0.25 ± 0.01)]. FAK treatment of these cells (S+FAK-I) decreased overall gene expression for these pro-inflammatory markers compared with S [CCL2 (0.13 ± 0.004, ▲p < 0.05 vs. 0.42 ± 0.009), IL6 (0.05 ± 0.002, ▲p < 0.05 vs. 0.28 ± 0.006)] (Fig. 5D).
We then repeated the multiaxial mechanical strain experiments on lung cells taken from patients with IPF and measured relative gene expression. Again, the pro-fibrotic genes CCN2, ACTA2, and TGFB1 increased in response to mechanical strain [CCN2 (1.32 ± 0.07, *p < 0.05 vs. 1.14 ± 0.06), ACTA2 (1.49 ± 0.008, *p < 0.05 vs. 1.27 ± 0.04) and TGFB1 (1.71 ± 0.04, *p < 0.05 vs. 1.48 ± 0.04)]. However, blocking mechanotransduction with FAK-I in (S+FAK-I) decreased CCN2 gene expression (0.98 ± 0.04, ▲p < 0.05 vs. 1.32 ± 0.07), but no significant difference was observed in ACTA2 (1.51 ± 0.01 vs. 1.49 ± 0.008) and TGFB1 (1.73 ± 0.02 vs. 1.71 ± 0.04) (Fig. 5E). Examining pro-inflammatory genes CCL2 and IL6 in IPF cells, we found that they also increased in response to strain [CCL2 (1.71 ± 0.05 vs. 1.67 ± 0.03), IL6 (4.89 ± 0.09, *p < 0.05 vs. 3.22 ± 0.06)]. However, this increase was only partially blocked with FAK-I treatment (S+FAK-I), [CCL2 (1.46 ± 0.02, ▲p < 0.05 vs. 1.71 ± 0.05), IL6 (4.80 ± 0.14 vs. 4.89 ± 0.09)] (Fig. 5F). Interestingly, CCN2 and CCL2, which are responsive to FAK-I, are involved in the accumulation of ECM at earlier stages in fibrosis. 34 These findings suggest that FAK inhibition could have a differential effect on early gene signals, downstream of mechanotransduction pathways.
To contextualize the differential effects of FAK-I on normal human and IPF fibroblasts, individual gene expression values in these treatment groups (n = 8) were normalized. Utilizing this approach, we demonstrated that FAK-I decreased the expression of pro-inflammatory and pro-fibrotic genes in both normal and IPF fibroblasts (Fig. 5G-H); however, the strength of this decrease was substantially stronger in normal human lung fibroblasts when compared with human IPF fibroblasts across a broad range of genes. This suggests that FAK-I may be less successful as a therapeutic in the end stages of IPF as seen in these explanted IPF lungs and may be more useful earlier in the disease course.
DISCUSSION
The IPF is a chronic, progressive lung disease that is characterized by scar tissue formation and interstitial pneumonia. 35 The exact etiology and pathophysiology remains unknown, and the patient outcomes remain grim. 36 Different groups have attempted interventions to mitigate progressive pulmonary fibrosis, but because of the multifaceted interactions of different pathways and cell types, a viable clinical therapy is yet to be developed. 37 –40 FAK-I has been clinically used in oncology and is undergoing evaluation for other disease indications such as fibrosis and scarring. 14,41 –44 Physiologic lung function requires constant stretch and contractile activity to move lung gases, 45 and this provides strain-induced activation of FAK in a variety of cells. 46 Myofibroblasts have been proposed as having a central role in pulmonary fibrosis, resulting in excessive deposition, contraction, and stiffening of the collagenous ECM. 47 In this study, we sought to further evaluate the role of FAK in mediating key components of the fibrotic and inflammatory response pathways in both normal and IPF cells, which may yield critical insights into the pathogenesis of IPF.
Since mechanotransduction seems to be important, 6 we targeted the FAK pathway to counter the IPF fibrotic response. The FAK-KO in vivo experiments corroborated the important role of FAK in driving a network of genes that are responsible for fibrosis. Knocking out FAK reduced the density of collagen networks, resulting in a less dense and less scar-like appearance of collagen fiber arrangement, as well as decreased pro-fibrotic expression of Acta2, Col1a1, and Tgfb1 genes that regulate the fibrotic response by mediating cellular contractility, mobility, and ECM deposition. 48 –52 These findings are corroborated within the literature. 53 In healthy and IPF human fibroblasts in vitro, we also found that increasing mechanical strain upregulated the expression of pro-inflammatory (IL6, CCL2) and pro-fibrotic (CCN2, ACTA2, TGFB1) genes, which drive collagen deposition, proliferation, inflammation, and angiogenesis. 54 –56 Interestingly, subsequent FAK inhibition decreased the expression of these genes only in normal fibroblasts, whereas IPF fibroblasts were less responsive.
Fibrogenesis pathways are robust and multifaceted, 6,57 contributing to the complexity and difficulty of treating IPF. To our knowledge, we are the first to evaluate and compare the importance of acute mechanotransduction on both normal and IPF human fibroblasts. The responsiveness of human IPF cells to FAK inhibition after mechanical strain was significantly less than in normal human lung cells, and these differentially favorable effects have not been previously identified in the literature. Our data suggest that targeting FAK at an earlier stage may lead to more beneficial outcomes compared with in later stage IPF. From our data, we suggest that FAKI therapy could work best during the early progression into IPF and be less effective on patients with chronic IPF.
Minimized response in chronic IPF fibroblasts to FAK-I could be due to an elevated amount of fibrotic and inflammatory genes natively present in IPF cells. Further, Nanchahal and Hinz described that late-stage fibrotic tissues are relatively acellular, with a few cells available to target in the densely cross-linked collagen matrix. 58 With less cells available for therapeutic intervention, the effectiveness of treating late-stage fibrosis could be minimized. In the fibroproliferative pathology of IPF, a pathologic matrix is progressively formed through the recruitment resident cells to overactivate an ECM repair process, resulting in fibrotic scar tissue that remains permanently present. 59 As different cells replace the ECM, the dysfunctional and inert scar tissue suppresses native organ function and induces stiffness, creating a vicious cycle that eventually leaves whole organ transplant as the only treatment option. Future work will be done to determine whether increasing the concentration of FAK-I therapy could potentially overcome these effects. 60
Overall, these data from our initial study confirm the importance of mechanotransduction in driving chronic IPF in both mouse and humans. Importantly, simultaneously targeting other pro-fibrotic genes, such as TGF-β or CCN2, to attenuate the fibrotic response can also be merited and requires further investigation. 61,62 Given the complexity of the fibrotic and inflammatory response pathways in IPF, administering FAK-I at early stages of fibrosis could have the greatest effects at attenuating the FAK-mediated fibrotic response pathway in IPF, 41,46,63 resulting in a more effective therapeutic solution that is suitable for clinical application.
CONCLUSION
In this study, we observed that FAK has a central role in mediating pro-fibrotic and pro-inflammatory pathways during IPF. Knocking out FAK reduced fibrosis and inflammation in an established acute model of pulmonary fibrosis, confirming the critical importance of mechanical strain in modulating the multifactorial pathophysiology of IPF in these models. We observed that early intervention in disrupting mechanotransduction pathways in the fibrotic response may enable the greatest reduction in IPF-associated changes in tissues and fibroblasts. FAK-I has shown great promise in other disease indications, outside of oncology, and merits further safety and efficacy studies. Overall, our study provides further insight into the interconnected role of FAK and the fibrotic and inflammatory response pathways within IPF that were observed in an acute model of fibrosis. Targeting early stages of fibrosis may yield beneficial clinical therapies for patients with IPF.
KEY FINDINGS
Mechanical strain significantly increases pro-inflammatory and pro-fibrotic genes.
The FAK plays a key role in mediating an interconnected network of pro-inflammatory and pro-fibrotic genes.
The FAK inhibition has a diminished effect on human IPF fibroblasts compared with normal human cells.
Early stage interventions may yield the greatest effects in attenuating fibrosis.
Footnotes
ACKNOWLEDGMENTS AND FUNDING SOURCES
The authors thank Theresa Carlomagno and Tiffine Vang for administrative support. This work was supported partially by the Hagey Family Program Funds.
AUTHOR DISCLOSURE AND GHOSTWRITING
No competing financial interests exist.
ABOUT THE AUTHORS
AUTHORs' CONTRIBUTIONS
A.A.T., K.M., and G.C.G. designed the study. A.A.T., K.C., S.H.K, D.H., D.S., C.A.B., and K.M. performed the animal and cell culture experiments. A.M.MB., S.M., S.R.S., and M.R.L. performed image analysis. A.A.T., K.C., D.H., M.J., and G.C.G. wrote the article. M.R., C.N., Y.J., J.P., D.P.P., M.C.L., G.G., H.C.K., A.C., M.J., D.C.W., K.S.F., and M.T.L. helped revise and edit the article.