
Abstract
Significance:
Increasing development of experimental animal models has allowed for the study of scar formation. However, many pathophysiological unknowns remain in the longest stage of healing, the remodeling stage, which may continue for a year or more. The wound healing process results in different types of scarring classified as normal or pathological depending on failures at each stage. Failures can also occur during wound remodeling, but the molecular mechanisms driving the wound remodeling process have yet to be investigated.
Recent Advances:
While the current understanding of wound repair is based on investigations of acute healing, these experimental models have informed knowledge of key components of remodeling. This review examines the components that contribute to collagen organization and the final scar, including cell types, their regulation, and signaling pathways. Dysregulation in any one of these components causes pathologic healing.
Critical Issues and Future Directions:
As wounds continue to remodel months to years after reepithelialization, new models to better understand long-term remodeling will be critical for improving healing outcomes. Further investigation of the contributions of fibroblasts and cell signaling pathways involved during remodeling as well as their potential failures may inform new approaches in promoting regenerative healing beyond reepithelialization.

Scope and Significance
Skin scarring causes significant physical, psychological, and financial burdens for patients. To date, there is no treatment to overcome skin fibrosis. This article aims (i) to outline the stages of wound healing, (ii) specifically, review the current state of knowledge in wound remodeling, and (iii) to discuss potential future study required to advance knowledge in wound remodeling.
Translational Relevance
This review examines the knowledge of cellular mechanisms driving wound repair and regeneration. Improved understanding of the underlying physiologic mechanisms that impact healing after reepithelialization may inform clinical practice around regenerative wound healing.
Clinical Relevance
After closure, wounds remodel for months to years leaving scars that never regain the strength of unwounded skin. Advances in knowledge of wound remodeling may improve management of skin scars, facilitating the healing of insults to the skin for maximal restoration and esthetic outcomes for patients.
Background
As the first barrier to external stress, including physical, biological, and chemical agents, skin is subject to frequent injury and tissue damage. Wounds are caused by insults involving the dermis, including surgical incisions, burns, and other traumatic injuries. While the skin has complex protective mechanisms, these insults to the skin yield fibrotic scars. Scar formation is the physiologic endpoint of the remodeling phase during wound healing.
Defining features of skin scars include the presence of dense extracellular matrix (ECM) and the absence of secondary features (hair follicles, sebaceous glands). 1 In optimal healing conditions, skin restoration that occurs as granulation tissue is remodeled to mature scar. Depending on the location, formation of normal skin scars can impair function. In suboptimal or “pathologic” healing conditions, “over-healing” due to excessive collagen synthesis can lead to hypertrophic scars or keloid formation. 1 Each year hundreds of millions of patients are affected by skin scars worldwide. 2
On the other end of the spectrum, “under-healing” during the remodeling phase results in loss of original tissue in the form of chronic wounds. Chronic wounds present a significant clinical challenge and are defined as the failure to reach healing endpoints or reepithelialization. 2 Many variables can impact wound healing and lead to impaired closure, including infection, nutritional deficiencies, ischemia/reperfusion injury, smoking, radiation, defects in collagen synthesis, or altered immune response. 3 These factors may be attributed to chronic or systemic illnesses, such as diabetes, and vascular and cardiopulmonary diseases, and result in ongoing inflammation and impaired blood flow. 4
Aging also contributes to compromised wound healing due to hormonal changes, such as estrogen deficiency, combined with increased rates of comorbid conditions that are influential in the attenuated wound repair. 5,6 The mortality rate for chronic wounds, such as diabetic foot ulcers or ischemic ulcers, is higher compared with breast or prostate cancer. 7,8
Treatments for both skin scars and chronic wounds remain a focus of research. Although the annual market for treatments is estimated at $12 billion, 2 existing therapeutic options are limited. There are no standardized treatment options for hypertrophic scars and while interventions exist for chronic wounds, more efficacious alternatives are needed. Physical management options are often considered for established scars to target the remodeling phase.
The main treatment approaches involve either silicone dressings or compression garments. Silicone gel sheets provide occlusive barriers, preventing water loss. 4 Silicone dressings are also thought to reduce tension at the scar border and reduce inflammation. 9 Compression garments are hypothesized to increase rigidity of the ECM and decrease mechanotransduction, which may alter fibroblast differentiation and proliferation. 9 A better understanding of the remodeling phase may allow for advancement in new and effective therapies for the management of both scars and chronic wounds.
As we continue to gain insights into wound healing mechanisms and specific cell contributions in wound repair, many pathophysiological unknowns remain in the remodeling phase of healing. One area of investigation in wound healing is the contribution of mechanotransduction. Following skin repair, scars thicken due to high skin tension. 1 As wound tension increases, profibrotic genes (type I collagen [COL1], transforming growth factor-beta [TGF-β]), are upregulated. Inhibition of mechanosignaling pathways (focal adhesion kinase inhibition) as well as physical offloading reduces scarring and allows for regeneration. 1 However, the role of mechanotransduction in wound remodeling remains unexplored.
During remodeling, the final stage of wound repair, granulation tissue is replaced as fibroblasts lay down collagen, which is remodeled from type III collagen (COL3) to COL1 to form mature scar. 9 The wound remodeling process may continue for well over a year, but the healed tissue never returns to a state that is comparable to uninjured skin. While tissue biomarkers have been studied for use in predicting healing impairment, 10 the molecular mechanisms driving the wound remodeling process have yet to be investigated.
Stages of Wound Repair
While wound healing involves a complex interplay between cell types, it is often described within the framework of its four major stages (Fig. 1). Healing proceeds through hemostatic and inflammatory responses, proliferation and cellular migration, and finally matrix deposition and remodeling. 11 These stages create a framework and can be used as a guide to define cell pathways involved in each phase of healing and increase understanding of impaired healing. While human skin wound healing follows a general timeline, the exact timepoints of each phase depend on additional factors such as size and complexity of the wound. 3,12 The timing of these stages also varies in acute versus chronic wounds.

Stages of wound repair. Wound healing is classically divided into four stages: (1) hemostasis, (2) inflammation, (3) proliferation, and (4) remodeling. Coordinated cellular response is key for wound repair. Each stage is driven by specific growth factors, produced by the cells depicted. 46 EGFs, epidermal growth factors; FGFs, fibroblast growth factors; HGF, hepatocyte growth factor; IGF, insulin-like growth factor; IL-1, interleukin-1; PDGF, platelet-derived growth factor; TGF-α/β, transforming growth factor-alpha/beta; VEGF, vascular endothelial growth factor.
Hemostasis
The initial phase of hemostasis starts immediately after tissue injury to stop vascular damage. Injury activates the intrinsic and extrinsic coagulation cascades. 13 Following collagen exposure, platelets aggregate to form a platelet plug. 14 Secondary hemostasis follows with activation of the coagulation cascade converting fibrinogen to fibrin monomers, creating a fibrin mesh. 10 Finally, a thrombus forms to allow for the cessation of bleeding and to serve as a scaffold for infiltrating cells during the inflammation and proliferation stages of healing. 14 Platelets also release growth factors and chemokines within the wound environment to stabilize the wound and to recruit macrophages and fibroblasts. 10
Inflammation
The second phase of wound repair is inflammation, which also starts at the time of injury. 10 Inflammation occurs and is characterized by the presence of edema and neutrophils within the tissue following histamine release and vasodilation through the complement cascade. 15 Acute inflammation is an immediate response to cell injury and activates the innate immune system for local and systemic defense. Toll-like receptors on skin cells are exposed to pathogen-associated molecular patterns, 16 upregulating nuclear factor kappa B to activate the immune response and production of immune mediators. 17 Inflammation after tissue injury also occurs by recognition of alarmins, or damage-associated molecular patterns, released by damaged cells. 10 Leukocytes, specifically neutrophils, migrate to the site of injury as a response to both vasodilation and chemotaxis signaling.
Neutrophils kill bacteria through oxidative burst mechanisms, including superoxide and hydrogen peroxide formation. 12 The acute phase response is replaced by macrophages after inflammation begins to clean the wound area through phagocytosis. 12 Macrophages simultaneously release anti-inflammatory cytokines (interleukin [IL]-10 and TGF-β) and attract additional neutrophils through IL-8. 18 Macrophages also stimulate fibrogenic growth factors and cytokines required for tissue repair, including platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF). 12
Proliferation
Fibroblasts are recruited, which are critical for phase 3 due to their role in ECM synthesis and remodeling. Proliferation is characterized by epithelialization and formation of granulation tissue. 12 Inflammatory cytokines, including IL-1, initiate epithelialization. Fibroblasts secrete keratinocyte growth factors (KGF-1, KGF-2), which along with IL-6, signal keratinocytes to migrate to and proliferate at the wound site. 12 Fibroblasts also produce matrix metalloproteinases (MMPs), which remove inflammatory debris and degrade damaged matrix to allow migration of new fibroblasts to the area of injury. These migrating fibroblasts produce ECM proteins to provide a provisional framework and allow for the formation of granulation tissue. Granulation tissue consists mainly of fibroblasts, which synthesize and deposit new collagen. 12
Neovascularization or angiogenesis within granulation tissues provides nutrients required for new tissue growth. IL-6 has also been shown to modulate TGF-β expression, inducing collagen production as well as fibroblast differentiation to myofibroblasts. 12 When the tissue defect is filled with granulation tissue, the wound margins contract the action of myofibroblasts to close the wound. Epithelialization is an essential component of skin healing and restores the wound barrier to protect against infection and fluid loss. 12
Remodeling
The final phase can last for over 2 years and involves remodeling of the ECM resulting in restoration of the architecture of the skin. 13 Tissue remodeling is characterized by the transition from granulation tissue to scar formation as COL3 is replaced by COL1 to increase tissue strength. 12 Remodeling of the wound bed continues, and further contraction takes place to strengthen the scarred tissue.
Current Animal Models to Study Wound Remodeling
Both small (e.g., murine) and large animal (e.g., porcine) models are commonly used in the study of wound healing. While there are limitations to these models as their skin anatomy differs from human skin, they remain the most suitable options and offer more than in vitro studies. Animal models allow for the study of similar biological environments and the relevant cellular and molecular interactions involved in both acute and chronic wound healing. The rodent model is a common animal model as it is relatively low cost and widely available. 19 One major difference is the panniculus carnosus muscle present in the subcutaneous layer in rodent skin, which facilitates rapid wound contraction. 19 Porcine models are also used as the skin is most like human skin, due to its thick epidermis and dermis and the presence of apocrine glands. 19 However, the major limitations of this large animal model are the high costs and poor vascularization in the dermis. 19
A variety of wound skin models have been established using both small and large animal models, including incisional, excisional, excisional splinted, ischemic, and infected wounds. 19 Burns, ulcers, and human to mouse xenografts have also been used to study mechanisms of dysfunctional healing and test therapeutic interventions. 19
Other techniques, such as lineage tracing and knockout mice, have allowed for the investigation of specific cell types and their roles in wound healing. 10 Lineage tracing is a way of studying cell contributions by identifying cells that express a certain protein using genetically encoded fluorescent labeling in transgenic mice. 19 Inducible Cre-lox systems, as well as constitutive Cre drivers, have been used to understand distinct phenotypes and cell differentiation from progenitor to mature states. Furthermore, the use of knockout mouse models allows for the study of cell function by testing healing outcomes when specific genes are not expressed. 10
Key Components of Wound Remodeling
Myofibroblasts in wound remodeling
Myofibroblasts are required for ECM reorganization and are a subset of fibroblasts. 20 The exact origin of myofibroblasts is unknown, however, multiple cell types may give rise to myofibroblasts. 21 After tissue injury, fibroblasts migrate to fill the wound bed resulting in granulation tissue and begin to exert mechanical force on the wound matrix. 22
Fibroblasts then differentiate into myofibroblasts due to continued increases in contractile force and synthesis of α-smooth muscle actin (α-SMA), the presence of which distinguishes myofibroblasts from their previous phenotypes and plays a role in their contractile function. 23 The expression of α-SMA directly correlates with the force generated by myofibroblasts. 22 Myofibroblasts also respond to TGF-β and PDGF signaling for ECM deposition to occur within the wound. 23 Ultimately, activation of myofibroblasts requires the TGF-β signaling pathways, mechanical stress, and myofibroblast maintenance molecules such as extra domain A-fibronectin and hyaluronan (Fig. 2). 23
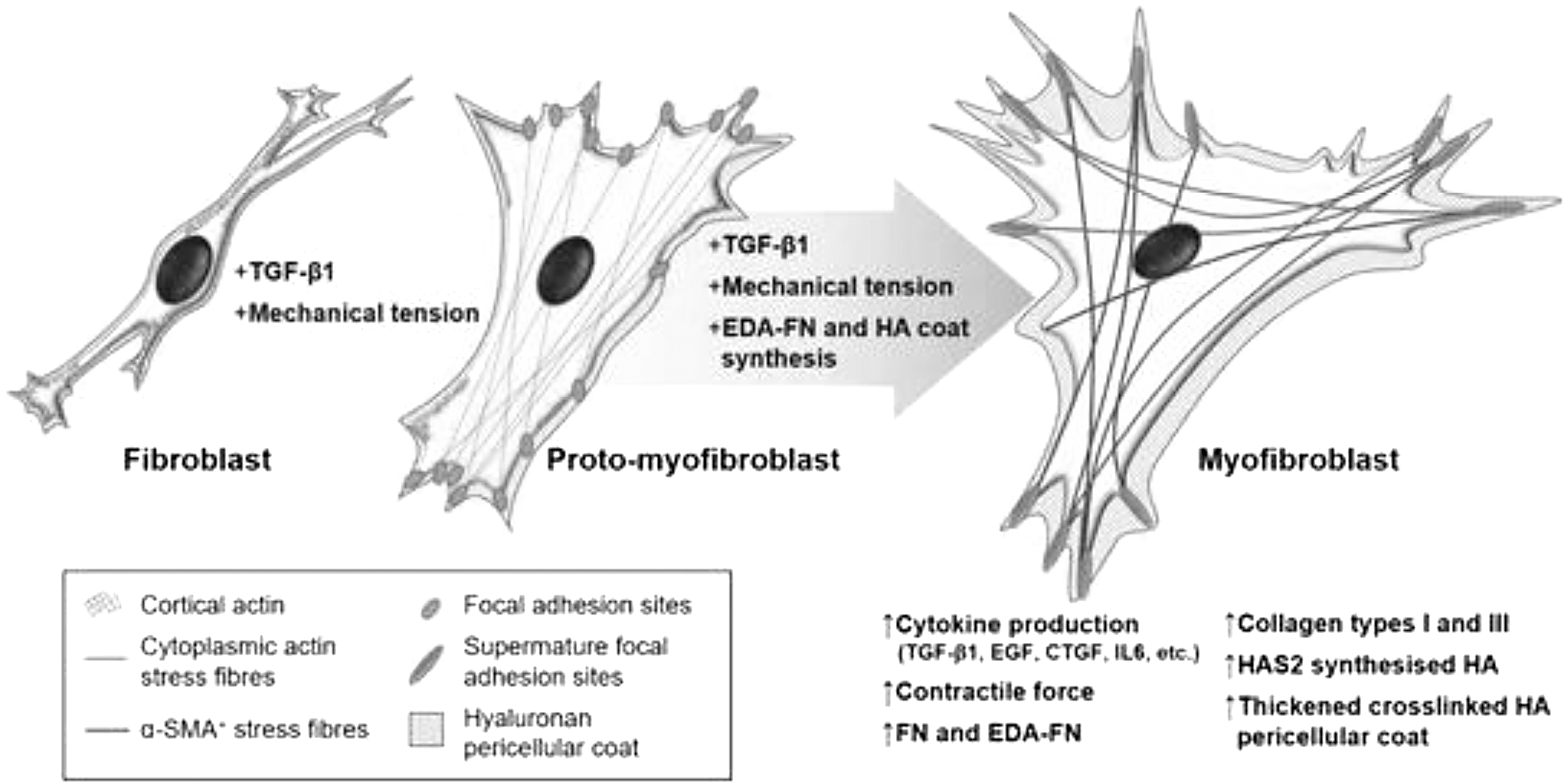
The process of fibroblast–myofibroblast differentiation. The activation of the TGF-β1/Smad signaling pathway, cell–ECM mechanotransduction signaling, and the synthesis of modulators maintain the myofibroblast phenotype. Current understanding is that myofibroblast differentiation is a continuum of different states, of which one intermediate cell type can be characterized as the “proto-myofibroblast.” This intermediate stage can be distinguished by increased proliferation, migration, and the rearrangement of cortical, membrane-associated actin into cytoplasmic filamentous actin stress fibers. The mature myofibroblast exhibits thick and crosslinked HA pericellular coats and contains α-SMA+ cytoplasmic actin stress fibers. ECM, extracellular matrix; EDA-FN, extra domain A-fibronectin; HA, hyaluronan; HAS2, hyaluronan synthase 2; α-SMA, α-smooth muscle actin. Taken with permission from Tai et al. 23
Therefore, remodeling occurs under the mechanical signaling of myofibroblasts as tension facilitates both TGF-β production and α-SMA expression in a positive feedback loop. 22 However, there is no clear understanding of the role of mechanics during the wound remodeling phase.
During remodeling, epithelial–mesenchymal transitions (EMT) regulate wound integrity and tissue homeostasis. 24 Myofibroblasts lay down collagen and other ECM elements. As healing processes activated after initial injury begin to slow, new collagen is continuously added for stabilization of the contracted matrix. 13 As this occurs, myofibroblasts can either undergo apoptosis or continue to contract by attaching to adjacent collagen networks, generating additional force required for shortening of the collagen matrix. Collagen crosslinking during remodeling is thought to cause increased stiffness and contracture. 22 However, future studies are required to understand the exact reassembly and formation of linked collagen fibers during remodeling. Due to the important role in wound healing, understanding the interactions of myofibroblasts and other cell types is important to intervene in wound repair.
MMP activity
MMPs, or “remodeling” proteinases, are endopeptidases and play critical roles in all stages of wound healing, including remodeling of the ECM. 22 Low levels of MMPs are expressed in normal tissue. When tissue remodeling is needed, MMP activation is rapidly induced. After reepithelialization, fibroblasts upregulated by TGF-β and IL-6, synthesize both MMPs and tissue inhibitors of metalloproteinases (TIMPs). However, several cell types can induce the expression of MMPs, including fibroblasts, keratinocytes, inflammatory cells (macrophages, lymphocytes), and endothelial cells. 25 The location and specific stage of wound repair determine which MMPs are expressed. 25 MMPs' main roles are to degrade existing ECM components, such as fibronectin. Matrix remodeling can also occur through intracellular degradation (endocytosis or phagocytosis) and allows for the migration of certain cell types, such as fibroblasts, required for wound repair. 26,27
TIMPs are endogenous inhibitors of MMP activity and prevent further modification of ECM. 12,28 Therefore, the roles of TIMPs are also important to consider during remodeling. The balance between collagenolytic activity and endogenous inhibitors is critical to create a stable matrix (Fig. 3). 3 Myofibroblasts manage this balance and can decrease MMP activity by upregulating TIMP production. 28 TIMPs therefore block additional ECM degradation. TIMP-3 has been shown to participate in ECM remodeling, but overall, the roles of specific MMPs and TIMPs are not fully understood. 25 Interestingly, higher ratios of MMPs to TIMPs have been observed in fetal wounds, known to heal with regenerative phenotypes, indicating the importance of fibrinolysis in healing. 10
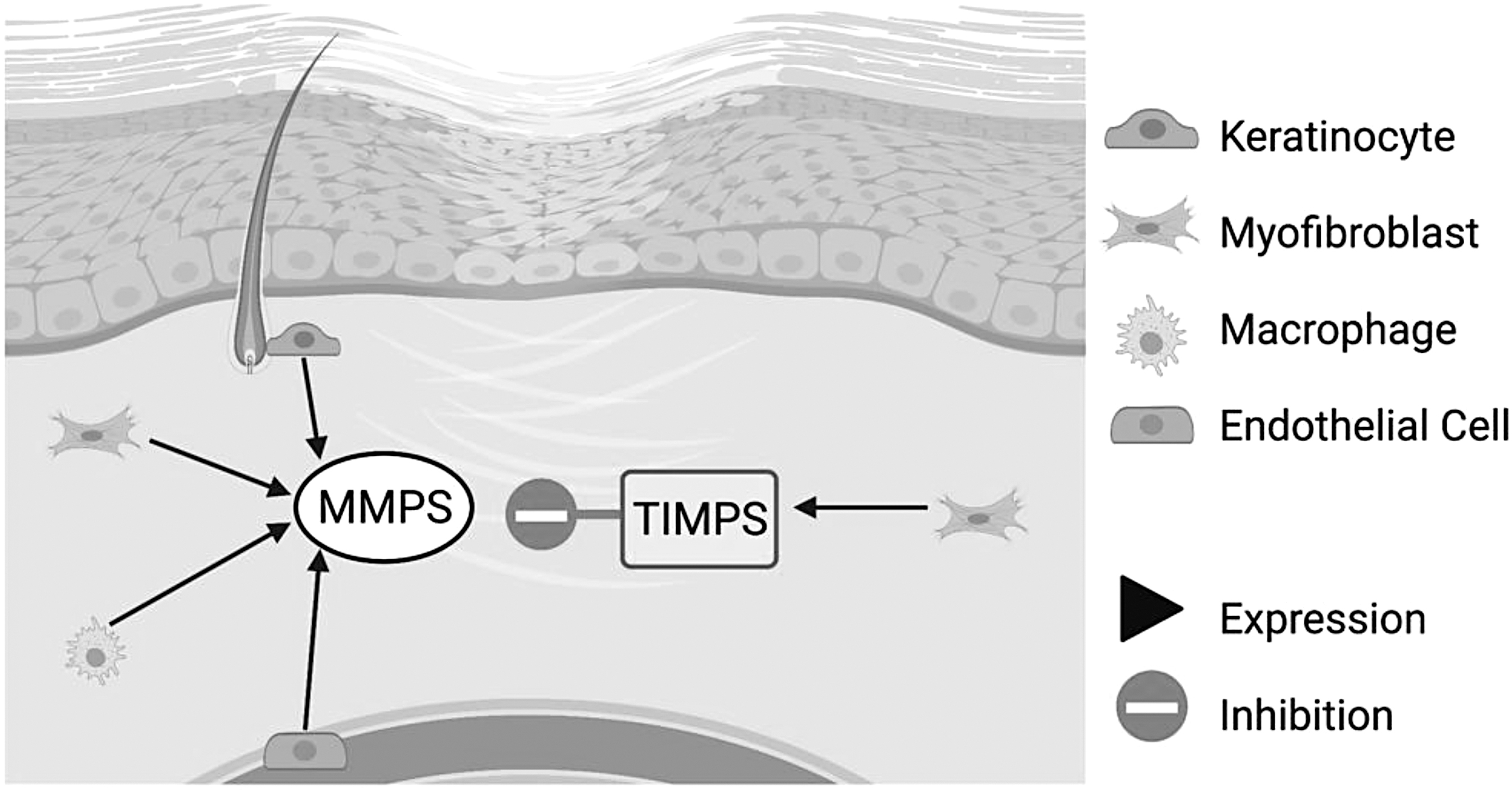
The role of MMPs in wound repair. MMPs are rapidly expressed in wound healing when tissue remodeling is required. Inflammatory cells (macrophages, lymphocytes), keratinocytes, fibroblasts, and endothelial cells can express MMPs in the skin. During proliferation, MMPs allow activated myofibroblasts to migrate into the wound area to lay down new ECM. Migration of epithelial cells allows for wound closure. After reepithelialization, TIMPs decrease promigratory MMPs to block additional ECM degradation. 28 MMPs, matrix metalloproteinases; TIMP, tissue inhibitor of metalloproteinase.
Regulation of ECM consolidation during remodeling is poorly understood, but murine models have explored the spatial relationships of collagen fibers after maturation. For example, Pensalfini et al. demonstrated that replacement of COL3 by COL1 starts at the lower region of the wound's center and that remodeling allows the mature wound to restore connections between the edges and the center of the wound bed. 29 Due to the importance of MMP activity during wound remodeling, the fine balance of MMP activity should be investigated further.
Growth factors in wound remodeling
Repair of tissue injury is mediated by paracrine signaling and includes many growth factors, including TGF-β, PDGF, and fibroblast growth factor (FGF) (Table 1). 30 These major growth factors are increased in acute wound healing and decreased in chronic wounds. 31 New matrix formation requires the transition from granulation tissue to collagen. PDGF mediates the removal of old collagen and TGF-β is involved in new collagen or ECM formation.
Growth factors in wound remodeling
While other growth factors may play a role in remodeling, these growth factors are reviewed in the literature.
Ang1, angiopoietin-1; ECM, extracellular matrix; FGF, fibroblast growth factor; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-beta.
Both PDGF and TGF-β are produced by fibroblasts, macrophages, keratinocytes, and platelets. 26 PDGF is an important growth factor throughout the stages of wound healing and plays important roles in blood vessel maturation and fibroblast proliferation. During wound remodeling, PDGF upregulates MMPs to break down existing collagen. 26 MMPs, under the control of various cytokines and growth factors, can also regulate inflammation. 15 TGF-β increases expression of fibronectin, collagen, and protease inhibitors and is a known wound-healing and fibrosis-promoting growth factor. 31
FGFs are produced by fibroblasts, endothelial cells, smooth muscle cells, and macrophages. FGF, specifically FGF-2, also plays a role in matrix formation and remodeling and stimulates collagenase production. FGF-2 has been shown to promote the migration of fibroblasts. 31 Despite an improved understanding of these signaling pathways involved in remodeling, a comprehensive picture of the roles many key growth factors play in wound remodeling remains to be determined.
Vascular maturation
One key factor during remodeling is vascular maturation to allow for successful wound healing. The neovascularization occurs during the proliferation phase and is driven by VEGF. 12 Angiogenesis produces disorganized and highly permeable vessels. Immature vessels are formed through vasculogenesis and angiogenesis but require maturation of the wall, consisting of endothelial and mural cells forming an ECM. 32 When the wound bed is fully granulated with immature vasculature, vessel maturation begins. During the maturation phase, these cells are recruited and patterned based on the needs of the local environment. 32 The majority of the neovasculature involutes at the final stage of wound resolution. 10,32
Vessel stabilization is driven by multiple factors. Using a murine model, Jain et al. describes four key factors involved in vessel stabilization: PDGF, TGF-β, endothelium-specific receptor tyrosine kinase (Tie2), and its ligand Angiopoietin-1 (Ang1), and sphingosine-1-phosphate-1. 32 Mural cells are the main source of Ang1, which stabilizes newly formed vessels. Tie receptors are also required for vessel stabilization. 32 Ang1 has been shown to rescue edema and hemorrhage in murine models and S1P1 promotes recruitment of mural cells. 32 In murine models, resolution factors, such as Sprouty2, which regulates capillary growth, and pigment epithelium-derived factor, responsible for vascular pruning, drive vessel remodeling. 33 Pericytes regulate vascular permeability and play an important role in the formation of stable vasculature. 33 While many studies exist to describe maturation in acute healing, processes driving longer maturation remain unknown.
Fibroblast heterogeneity
Fibroblasts are critical in supporting normal scar formation following skin injury. Lineage tracing, tracking cells using transgenic mice, has led to a better understanding of fibroblast heterogeneity. 34 Fibroblasts range in function depending on the location in the body, but even in one tissue location, the fibroblast populations and their functions are diverse. 21 Within the skin, spatial segregation, defined by position relative to the epidermis, and marker identification have been used to explore fibroblast subpopulations.
Two distinct fibroblast lineages are defined within the dermis, the papillary and reticular fibroblast subpopulations. Papillary (superficial) and reticular (deep) dermis layers can be distinguished by cell surface markers. 35 As we continue to learn about fibroblast subpopulations, the role of fibroblast heterogeneity in wound remodeling remains largely unknown. Papillary and reticular fibroblasts serve an important role in wound repair, but further investigation of their specific cell signaling pathways involved in remodeling will enhance our understanding of healing after closure.
Hair follicle formation
One of the major characteristics of wound regeneration is hair follicle formation. After acute injury, stem cell populations in the hair follicle bulge, the deepest part of the permanent hair follicle, migrate toward the center of the wound to transiently amplify healing. 36 These stem cells can also induce activation of myofibroblasts or differentiation into smooth muscle cells. 37 In newly closed murine wounds, hair follicles are absent as dermal regeneration starts with cells of the reticular dermis, which cannot produce hair follicles. 21 At reepithelialization, dermal papilla fibroblasts at the base of the hair follicle activate Wnt/β-catenin to drive hair growth and arrector pili muscle formation. Wnt signaling activates the formation of new hair follicles within the wound (Fig. 4). Specifically, β-catenin activation in murine fibroblasts decrease hair follicle regeneration, while ablation promotes regeneration. 38

Diagram to illustrate hair follicle regeneration. Dermal papilla fibroblasts can signal unipotent stem cells in the hair follicle through the Wnt/β-catenin pathway to regenerate various cell types within the epidermis. 8 In return, the epidermal stem cells in the hair follicle bulge (*) can signal the fibroblasts to convert to myofibroblasts expressing α-SMA to help in wound contraction. 48 A subset of myofibroblasts can also become adipocytes, a transition that has been found to reduce scar formation. 34
Ongoing Wnt signaling in murine wounds not only promotes formation of hair follicles, but also plays a role in ECM remodeling, as collagen is replaced by subtypes present only in uninjured or newly developing skin. 39 Wnt signaling activates expression of Sonic hedgehog by murine epidermal cells resulting in proliferation of papillary dermal fibroblasts and ECM remodeling. 40 The complexity of hair follicle formation demonstrates the plasticity of the dermis during remodeling.
Failures of Wound Remodeling
This review highlights the key contributors to remodeling at the cellular level. Dysregulation in any one of these components causes pathologic healing (Fig. 5). In normal scars, the final scar depends on the organization of collagen. Abnormal collagen morphology or crosslinking is seen in hypertrophic scars. If myofibroblasts persist and lead to ongoing ECM remodeling, excessive wound contraction affects the collagen matrix and wound contraction evolves to contracture causing hypertrophy. 22 During remodeling, COL1 replaces COL3 to strengthen the skin and breaking strength increases from 3% at 1 week to 80% at 3 months after wounding. 3 However, the integrity of the remodeled wound site remains inferior to healthy skin. 41
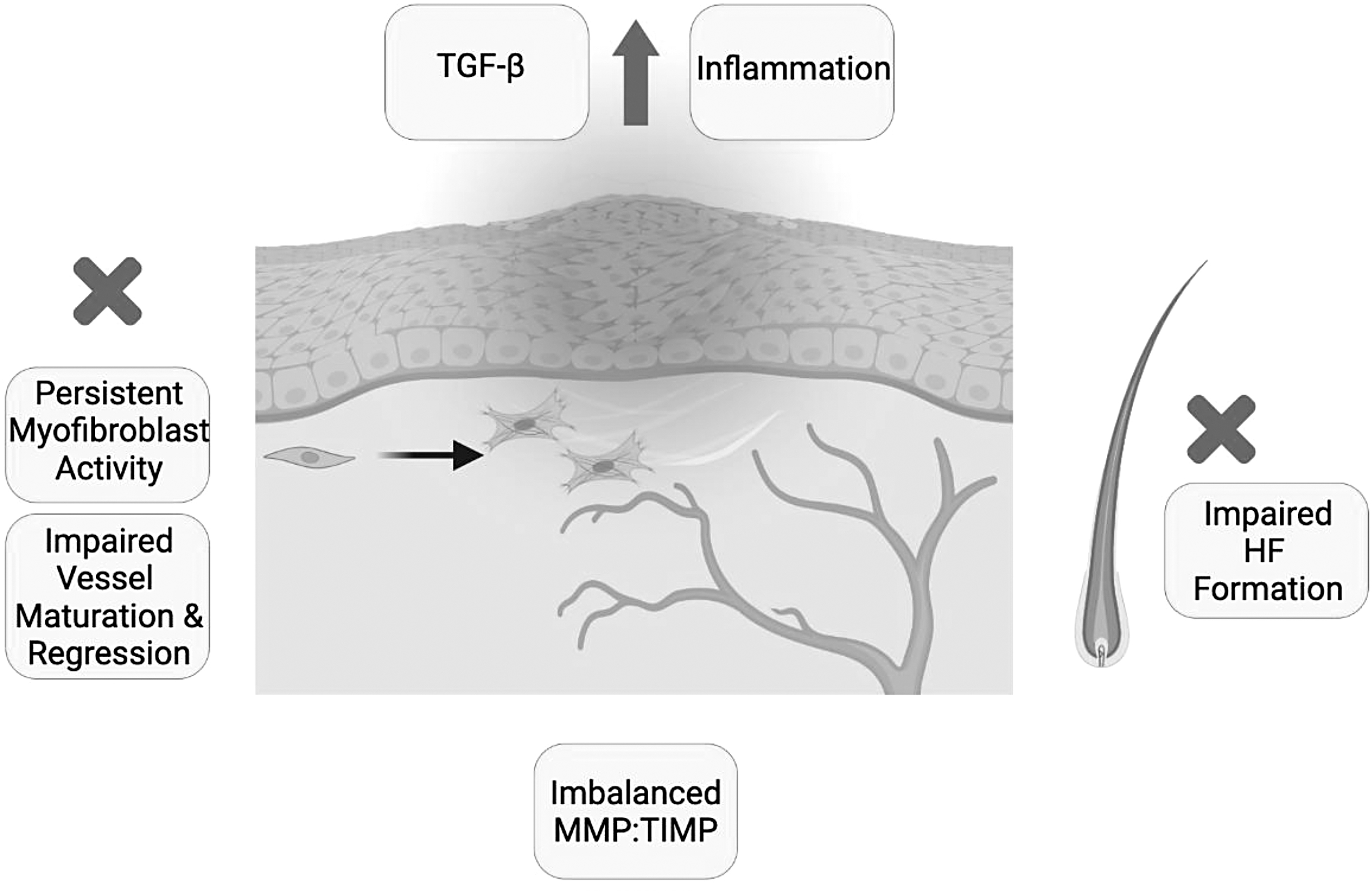
Failures of matrix remodeling. Dysregulation of key components that contribute to collagen organization and the final scar leads to failures during the remodeling stage. These include persistent myofibroblast activity 18 and impaired regression of neovasculature or vessel maturation. 27 Other areas of cellular dysfunction include imbalances in MMPs and TIMPs 9 and impaired HF formation. 40 Disrupted signaling pathways such as upregulated TGF-β or ongoing inflammation can also lead to pathologic healing. 45 HF, hair follicle.
Cellular dysfunction is one area causing failures in wound remodeling. Macrophages utilize a control mechanism to determine the level of myofibroblasts during remodeling. To prevent phagocytosis, myofibroblasts express CD47. However, if CD47 is overexpressed, myofibroblasts escape removal, which can lead to the formation of hypertrophic scarring. 10
Pericytes or excess endothelial cells causing microvascular occlusion can also contribute to failure of wound remodeling. 42 In addition, if papillary fibroblasts are decreased in capacity, hair follicles may not form as fibroblasts supporting hair follicle formation differentiate from the papillary fibroblast lineage. 43 Failure of reticular fibroblast formation will also cause abnormal wound remodeling as they determine reticular dermal and hypodermal formation. 44 Additionally, imbalances in MMP and TIMP levels are seen in nonhealing wounds. 45 Therefore, quantitative assessments of MMPs have been used to predict healing outcomes as TIMPs inhibit MMPs to prevent abnormal ECM degradation. 46
Remodeling is also affected by disrupted signaling pathways. Insufficient levels of growth factors essential for regulation of remodeling are observed in nonhealing wounds due to the absence of signaling molecules or atypical receptor locations. 31 The amount of TGF-β, for example, is increased in states of excessive healing, such as hypertrophic scar, as opposed to regenerative tissue. 13 While the role of EMT in wound healing is not fully understood, tumor necrosis factor-alpha has been shown to induce this transition and if not regulated, fibrosis can ensue. 10 The current knowledge of “normal wound remodeling” is limited and requires further investigation. Understanding its failures and how they can contribute to abnormal healing remains an area of critical importance.
Next Steps
While the early stages of wound healing are clearly delineated in the literature, many questions remain unanswered regarding the remodeling phase. Myofibroblasts are the key components, but their exact origin in remodeling remains unknown as they can derive from a variety of cells. The fate of these cells requires further investigation. In normal wound healing, these cells undergo apoptosis when the wound is closed, but they can also remain in the wound bed to continue ECM remodeling. 13
Newer research focused on antifibrotic pathways has targeted ways to inhibit myofibroblast activation or accelerate myofibroblast apoptosis. 47 It will be important to characterize the long-term role of fibroblasts in wound contraction, to prevent excessive healing. Additionally, given the fibroblast interaction with other cell types, understanding the fibroblast contribution to hair follicle formation and vascular maturation may present opportunities to promote regenerative healing.
Keratinocytes, for example, have been shown to respond to mechanical strain and modulate fibroblast proliferation and therefore remodeling, but the exact mechanisms are yet to be determined. 10 Additionally, much of our current understanding of vessel maturation comes from investigations of acute healing, but studies examining longer maturation time points and cellular contributions will guide future interventions.
Finally, the discovery of fibroblast heterogeneity has improved our understanding of the variety of fibroblast functions and efforts have also been made to induce the transition to proregenerative subtypes. 35 However, while we have gained understanding of the roles of fibroblasts during the early stages of healing, 48 the contributions of fibroblasts and cell signaling involved during remodeling remain at the levels of speculation. As wounds continue to remodel months to years after reepithelialization, new models to better understand remodeling will be critical for improving healing outcomes.
Summary
Hundreds of millions of patients each year are affected by skin scars worldwide. While billions of dollars are spent annually on wound care and scar treatments, little is known about the longest stage of healing, the remodeling stage, which may continue for a year or more. During remodeling, the scarred area is reinforced by COL1 but does not repair to the full strength of uninjured skin. Dysregulation in the remodeling process can cause pathologic healing, such as wound contracture or hypertrophy. Some key components have been identified allowing for better understanding of possible failures, which could be targeted to improve healing.
Take-Home Messages
Wound remodeling is the final and the longest stage of wound healing.
Little is known about the molecular mechanisms driving the wound remodeling phase.
Identifying causes of pathologic remodeling provides opportunities to improve wound healing and overcome scarring.
Footnotes
Authors' Contributions
A.F.S. completed the writing—original draft, investigation, and visualization. A.F.S. and M.F.G. designed the review, including conceptualization and methodology. M.F.G., D.C.W., and M.T.L. reviewed and edited the writing and validated the final author. All authors reviewed the final draft.
Acknowledgments and Funding Sources
The authors thank the Hagey Laboratory for Pediatric Regenerative Medicine for its continued support of our research. This work was supported by the Hagey Laboratory for Pediatric Regenerative Medicine.
Author Disclosure and Ghostwriting
The authors are researchers and have no conflicts of interest to declare.
About the Authors