
Abstract
Objective:
Biofilm infections in chronic wounds are common and pose a significant clinical challenge. This challenge was addressed by developing the SoftOx Biofilm Eradicator (SBE) composed of hypochlorous acid (HOCl) and acetic acid with strong broad-spectrum antimicrobial activity.
Approach:
First-in-human study investigating the safety and tolerability as primary endpoints and wound size effect and antimicrobial efficacy as secondary endpoints of SBE treatment in chronic leg wound patients. The study was divided into two as follows: a randomized, double-blinded, Single Ascending Dose (SAD) phase (n = 16 SBE; n = 4 placebo), where patients were treated with SBE or saline (placebo) only once, followed by an open-label, Multiple Ascending Dose (MAD) phase (n = 8), where patients were treated with SBE once daily or twice daily over five days. Reporting is according to CONSORT guidelines.
Results:
SBE was safe and well-tolerated in chronic leg wound patients. There were no significant differences in pain during and after treatment with SBE or the placebo. The SBE treatment reduced bioburden in wounds compared to baseline, with 98% and 49% median reduction after SBE or placebo treatment, respectively. A dose-dependent trend in absolute wound size reduction was observed in the MAD groups with a median (min, max) change of −2.99 (−14.25, −1.5) cm2 in the once-daily and −10.48 (−17.95, −0.38) cm2 in the twice-daily group, respectively.
Innovation and Conclusion:
This study demonstrated the safe use of HOCl-based SBE in chronic leg wounds with promising trends of immediate antimicrobial action and beneficial effect on wound healing.

INTRODUCTION
The prevalence of chronic wounds in the Western World is estimated to be as high as 2%, 1,2 and infection is one of the most frequently encountered complications of these chronic wounds. Once the protective skin barrier is compromised, all wounds become susceptible to invasion by a wide range of microbes, including bacteria, yeast, and fungi, originating from the environment, the surrounding skin, or the internal organ system. 3 A common characteristic of wound infections is the ability of the involved bacteria to protect themselves by forming biofilms in wounds, 4 which makes wound infections unresponsive to conventional antibiotic treatments, thereby posing a significant medical challenge. 5
Biofilms are clustered microbial communities embedded in a protective slime-like matrix and they are extremely tolerant toward antibiotics and the defenses of the host immune system. 5 –7 There is growing evidence showing the presence of biofilms in chronic wounds and their adverse role in delaying normal wound healing. 4,5,8 –10 Current approaches for managing infections in wounds are not effective in eradicating biofilms without having adverse effects on the host. 5 Furthermore, inappropriate use of antibiotics to treat infections in wounds and their inability to eradicate biofilms in these wounds place patients at high risk for acquiring antibiotic-resistant organisms.
SoftOx Biofilm Eradicator (SBE) has been developed to address the limitations of currently available treatment options for chronic wounds, which present a large unmet medical need. 11,12 It is a water-based formulation containing sodium hypochlorite (NaOCl) in an acetic acid/sodium acetate buffer. At the pH value of SBE (pH = 4.3), sodium hypochlorite is predominantly present in the form of hypochlorous acid (HOCl). SBE is composed of HOCl at concentrations of 500–1000 µg/mL and acetic acid (HAc) at concentrations of 1–3% (w/w), both of which are naturally occurring molecules and have a long history of safe use in medicinal products and in solutions approved as medical devices. 13 –15 Both molecules exhibit broad-spectrum antimicrobial activity at the concentrations present in SBE. 16 –18
HOCl is used by the innate immune system as it is highly active against invading bacterial, viral, and fungal pathogens. 18 –22 Moreover, HOCl is active against biofilms, 14,23,24 and several studies suggest that it increases oxygenation of the wound site to improve healing. 25,26 The antimicrobial effect of HOCl is rapid and powerful by unspecifically reacting with and disrupting the function of key microbial molecules such as proteins, lipids, and nucleic acids, while remaining safe to mammalian cells and not promoting the emergence of new resistant microbes. 18,20,27
The use of HAc for treating infections was described almost 100 years ago for the treatment of military wound infections, where a 1% solution of HAc was shown to eliminate Pseudomonas aeruginosa from military injury wounds within two weeks. 28 Since then, several studies have been conducted showing the efficacy of HAc as a topical treatment agent, especially against dormant bacteria found in chronic infections. 13,29 –36 More importantly, recent studies have shown that antibiotic-resistant biofilms of problematic pathogens, such as P. aeruginosa and various species of mycobacteria, were rapidly killed by HAc. 16,37 –39 In SBE, HAc functions as a pH buffer to stabilize the HOCl, and together, they maximize the antimicrobial effect of SBE.
The primary objective of this clinical study was to assess the safety and tolerability of single and multiple doses of topically applied SBE in patients with chronic leg wounds. The secondary objective of the study was to assess changes in bacterial burden and wound size/healing in the leg wounds after treatment with SBE or saline. The data from this study will guide the identification of safe doses of SBE to be administered to subjects with chronic leg wounds in larger clinical studies.
INNOVATION
HOCl is a well-known broad-spectrum antimicrobial; however, due to the volatile nature of the molecule, wound treatment with HOCl is difficult. Furthermore, HOCl has a positive effect on skin barrier function and wound healing. 24 –26,40 In SBE, a patented technology makes it possible to stabilize the HOCl, and, in this study, we show both good safety and tolerability of SBE treatment of chronic leg ulcers and also positive impact on wound bioburden and wound size, parameters needed for a successful wound closure (Fig. 1).

First-in-human trial: Wound treatment with stabilized HOCl. HOCl, hypochlorous acid.
CLINICAL PROBLEM ADDRESSED
Chronic leg ulcers are an increasing challenge in the Western World, and treatment regimens are often based on personal opinion, as no effective or gold standard cure exists. Treatment normally focuses on addressing underlying causes, compression/bandaging, topical antimicrobial intervention, and often also systemic antibiotics. With an effective topical antimicrobial treatment as SBE, the need for systemic antibiotics might decrease, which should diminish further resistance development.
MATERIALS AND METHODS
Study design
Single-center clinical study investigating the safety and tolerability of randomized, double-blinded, placebo-controlled ascending single doses of topically applied SBE in patients with chronic leg wounds and of open-label once daily and twice daily dosing of topically applied SBE for five days in patients with chronic leg wounds. The study was sponsored by SoftOx Solutions AS (Fornebu, Norway) and was conducted at Dan Trials Aps, Zelo Phase 1 Unit, located at Bispebjerg Hospital (Copenhagen, Denmark) between May 2021 and August 2022. The study was approved by the Research Ethics Board (US HHS# IRB00003245) and the Danish Medicines Agency (EudraCT no.: 2021-000314-42). Since this study was conducted in cooperation with, and with the support from the Navy of the United States of America (USA), the clinical study protocol was reviewed and approved, and the study was monitored by the U.S. Army Medical Research and Development Command (USAMRDC) Office of Human Research Oversight (OHRO, previously HRPO, Log Number E02082.1a). The study was performed in accordance with the national regulations and the Declaration of Helsinki. Informed consent was obtained from all study subjects before any study-related activities were performed. The study is here reported according to CONSORT guidelines.
Study arms and treatment groups
The study was divided into two phases, Single Ascending Dose (SAD) and Multiple Ascending Dose (MAD) phase. The patients included in the SAD phase were treated with either single ascending doses of SBE or the placebo in a double-blinded manner. This SAD phase consisted of four groups (Group 1–4) with five patients in each (four patients treated with SBE and one patient treated with the placebo) and aimed to identify the highest tolerated dose of SBE in a randomized, double-blinded, and placebo-controlled manner with sequential evaluation of four single ascending doses of SBE. Before dose escalation, the blinded results were evaluated by the Safety Monitoring Committee (SMC). The second phase of the study (MAD phase) contained two groups (Group 5–6) with four patients in each, all treated with SBE once daily (OD) or twice daily (BID; bis in die) for five consecutive days and aimed to evaluate the safety and tolerability of multiple dosing of SBE. The investigators were not blinded in the MAD phase. The MAD phase tested different dosing regimens with the SBE formulation determined by the SMC based on the safety and tolerability of the different SBE formulations evaluated in the SAD phase of the study.
Subjects eligible for dosing in the single-dose groups were assigned to treatment (SBE or placebo) according to a computer-generated randomization list. The randomization took place at the last practical time before administration of investigational medicinal product (IMP) and was documented by the allocation of a randomization number.
IMP, SBE, consisted of HOCl at concentrations of either 500 µg/mL or 1000 µg/mL and acetic acid (HAc) at concentrations ranging from 1% to 3% (w/w), depending on the treatment group (Table 1). Sterile isotonic saline solution was used as the placebo.
Doses and dosing regimen for the assessment of SBE treatment in chronic leg ulcers
SBE, SoftOx Biofilm Eradicator; HOCl, hypochlorous acid; HAc, acetic acid; OD, once daily; BID, bis in die (Latin), twice daily.
SBE Dose Selection and escalation
The minimum inhibitory concentration (MIC) and the minimum bactericidal concentration (MBC) of SBE for five clinically relevant pathogens were determined to be 25 µg/mL HOCl + 0.25% HAc and 50 µg/mL HOCl + 0.50% HAc, respectively (data not shown). However, the in vitro experiments using wound biofilm models, where organic material (e.g., serum and blood) was present in the growth/treatment medium, showed the necessity of higher concentrations of HOCl and HAc to effectively kill biofilms (data not shown), suggesting that the presence of organic material in leg wounds can also inactivate much of the applied HOCl in SBE. It was therefore hypothesized that higher concentrations of HOCl and HAc (to stabilize the elevated levels of HOCl) would be required for effective killing of microorganisms in leg wounds in the current study. However, since the concentrations required for real-life conditions had not previously been determined, the starting dose in the current study was selected based on toxicity data rather than pharmacological data. The results from a previous safety and tolerability study in minipigs showed that daily administration of SBE up to 1000 µg/mL HOCl + 3% HAc was well tolerated (data not shown).
Considering the NOAEL (no-observed-adverse-effect level) value of 1000 µg/mL HOCl + 3% HAc that was obtained in the 28-days repeated-dose toxicology study in minipigs, the starting dose in the current study (i.e., 500 µg/mL HOCl + 1% HAc) provided a safety factor of 2 for HOCl and a factor of 3 for HAc. It is important to note that in the current study, SBE was in contact with the wounds for a shorter period compared to what was applied in the earlier mentioned in vivo toxicology study in minipigs (data not shown).
Based on these earlier in vitro and in vivo studies, we decided to use a single dose of SBE at 500 µg/mL HOCl + 1% HAc as the starting dose in the first SAD group. The dose of SBE was limited to 1000 µg/mL HOCl + 3% HAc, since this was the maximum evaluated and tolerated dose in the in vivo animal toxicology study. The concentrations of SBE used for the six groups are displayed in Table 1.
Inclusion and exclusion criteria
Patients were eligible to participate in this study if they were ≥18 years of age and suffered from a chronic leg wound (present for at least four weeks) with a size of 1 to 100 cm2. Patients were not eligible to participate in this study if they had clinically significant reduced perception of sensation or pain in the proximity of the wound, as assessed through Mechanical Detection Threshold and the Mechanical Pain Threshold, if there was necrotic tissue or cancer in the wound, if they had a pain score from the wound more than 40 mm (on a 0–100 mm VAS), or if they had severe ischemia in the same leg as the wound, as measured by the ankle brachial index, with values below 0.5 indicating severe ischemia.
Interventions and treatment regime
IMP, SBE, was provided to the clinical trial site in a 250 mL polyethylene terephthalate bottle, with high-density polyethylene caps, each containing 250 mL of SBE at the following concentrations: 500 µg/mL HOCl and 1% HAc, 500 µg/mL HOCl and 2% HAc, 500 µg/mL HOCl and 3% HAc, or 1000 µg/mL HOCl and 3% HAc. Sterile isotonic saline (Irriflex®, from Fresenius Kabi), containing 120 mL NaCl with a concentration of 9 mg/mL, was used as the placebo. The specific SBE concentrations used in each group are presented in Table 1.
In regard to blinding, the IMP (SBE or placebo) was delivered to the treating staff in a polypropylene gallipot, and both the blinded staff and the patient wore nose clips due to SBE’s distinct odor of HAc and chlorine. These nose clips were worn until the 20-min soaking in IMP, wound irrigation with sterile saline and dressing was done, and after ventilation of the room with an open window for a couple of minutes.
As a precaution, sentinel and staggered dosing were applied in the SAD groups. The safety of two patients treated on two different days (at least one of whom was treated with SBE) was reviewed before commencing dosing of the remaining patients in a SAD group (sentinel dosing), with an interval of at least 1 hour between the dosing of different patients (staggered dosing). The patients in MAD groups were treated in parallel.
A flowchart of the treatment procedure of the wounds with SBE or the placebo is shown in Figure 2. Briefly, following debridement (if found necessary by the clinician) and irrigation of the wound with isotonic saline, a sterile gauze dressing thoroughly soaked in 100–150 mL SBE or the placebo was placed on the wound without any rubbing of the wound. The wound, together with the SBE- or placebo-soaked gauze dressing, was wrapped in plastic stretch film and left for 20 min. After the treatment, the plastic stretch film and the gauze dressing were removed, and the wound was irrigated with isotonic saline and covered with an appropriate neutral foam wound dressing specified and prescribed by the treating clinician.
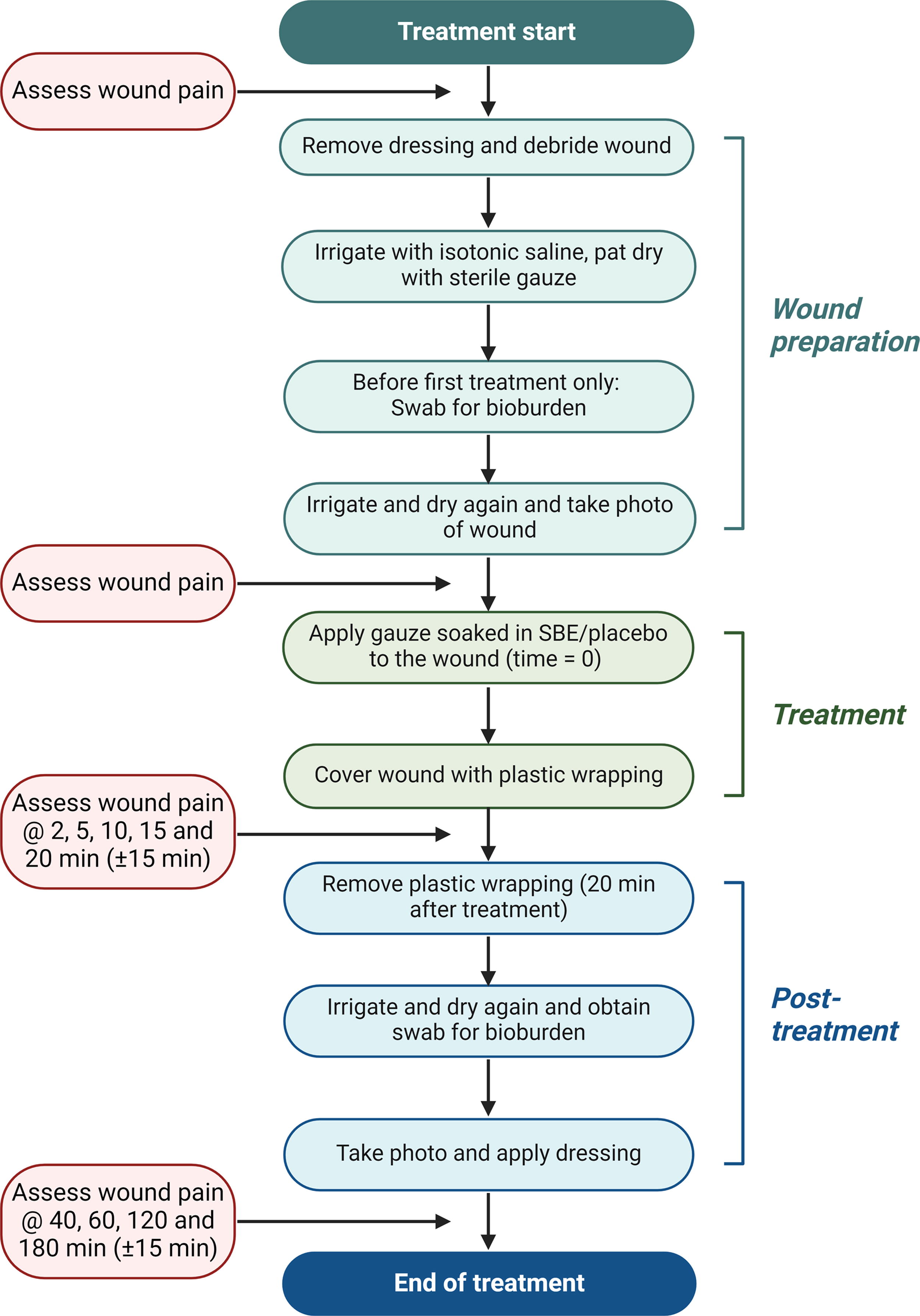
Flow diagram of wound treatment and pain assessment of patients with chronic leg ulcer.
Debridement was done at the discretion of the practitioner using forceps and/or with a curette and rinsed with saline.
The patients in the SAD groups attended three visits at the clinical trial site, a screening visit, a treatment visit, and a follow-up visit for final safety assessments 2–4 days after the treatment. The patients in the MAD groups attended seven visits at the clinical trial site: a screening visit, five treatment visits, and a follow-up visit for final safety assessments 2–4 days after the last treatment. Furthermore, for the patients in the MAD groups, a follow-up telephone call for safety assessment was scheduled 5–9 days after the last treatment.
SMC evaluated the blinded data for safety and tolerability of the treatment in all SAD and MAD groups prior to the initiation of the treatment in the next group. The SMC was able to request a more detailed analysis and/or unblinding of the data before deciding on the dose escalation or stop of the dose escalation.
Endpoints
The primary endpoints in the study were tolerability (wound pain) and the incidence and severity of adverse events (AEs) associated with the SBE treatment. Clinically significant abnormal values of vital signs (systolic and diastolic blood pressure, pulse rate, respiratory rate, and body temperature), safety blood and urine parameters, electrocardiogram (ECG), and physical examination were to be reported as AEs, if occurring within the AE reporting period. The secondary endpoints were the change in bacterial burden (number of colony-forming units per mL; CFU/mL) in the leg wound between the first administration of SBE or the placebo (the baseline) and after the last administration of SBE or the placebo and the change in wound size.
Variables
All AEs were registered from the time point of informed consent until the final study visit. The measurements carried out for the assessment of safety, tolerability, and efficacy of SBE treatment in SAD and MAD groups are listed in Supplementary Table S1 and S2, respectively.
Wound pain was measured using a visual analog scale (VAS) ranging between 0 and 100 mm, where 0 mm indicated no pain and 100 mm indicated the worst imaginable pain at the time of assessment. Wound pain was assessed at regular time intervals before, during, and after the administration of SBE or the placebo (Fig. 2). In addition, pain originating from the wound was used for excluding patients who had a wound pain score above 40 (based on a 0–100 mm VAS) already prior to the first treatment.
The presence of bacteria (bioburden) was assessed in the wounds prior to and after the treatment procedure and at the follow-up visit in the SAD groups and prior to the first treatment, after the last treatment on each day, and at the follow-up visit in the MAD groups. A sterile ESwab was gently stroked in a zigzag manner across the wound and was used for culturing and subsequent counting of CFU. The analysis was carried out at an external laboratory at the Costerton Biofilm Center, University of Copenhagen.
Wound size was calculated as the area of the wound in cm2 (the width X the length of the wound). The calculated wound size was used to exclude patients with a wound area at screening of <1 cm2 or >100 cm2. In addition, wound size was used for an exploratory assessment of any effect of the SBE treatment on wound closure. Wound size was assessed by acquiring images of the wounds at screening, at baseline, i.e., before the (first) administration of SBE or the placebo, and at the follow-up visit.
Other safety measurements, such as vital signs (blood pressure, pulse rate, respiratory rate, body temperature), blood and urine parameters, and ECG, were also carried out at single time points or at certain intervals (Supplementary Tables S1 and S2).
Microbiological analysis
Upon arrival at Costerton Biofilm Center, each swab sample was vortexed vigorously and sonicated in an ultrasound bath before 10-fold dilution series were performed in 0.9% (w/v) NaCl. From each dilution, 100 μL was plated onto 5% blood agar plates and blue agar plates (a modified Conradi–Drigalski medium) (Statens Serum Institute, Copenhagen, Denmark), and the plates were incubated at 37°C for 24 h. The CFU were counted to estimate the bacterial load (CFU/mL). Electronic laboratory notebook was not used.
Statistical analyses
No formal sample size calculation was performed, as the study had an exploratory nature; however, with a total of 24 subjects treated with SBE there was a probability of 0.77 that at least 1 adverse event (AE) of a certain kind would occur given a true common AE probability of 0.06 for that AE. All statistical analyses were performed using SAS® (Version 9.4 or higher, SAS Institute Inc., Cary, NC, USA). The data are presented by dose group, and the data for patients, who received placebo, were pooled across the treatment groups. The patients were analyzed by the dose they received. All the patients that were treated at least once with SBE were accounted for and were to be used in the following data analysis sets: Safety Analysis Set (SAS), which included all the patients that were treated at least once with SBE and for whom safety data were available. The patients were analyzed by the dose they received. The SAS was the primary analysis set for the safety analyses. Full Analysis Set (FAS) consisted of all patients that were randomized into the study. The FAS followed the intention-to-treat (ITT) principle, i.e., patients were analyzed in the treatment group to which they had been randomized, regardless of whether treatment was received as planned. The FAS was the primary analysis set for the analyses of efficacy.
Number of observations (n), mean, quartile 1 (Q1), median, quartile 3 (Q3), standard deviation (SD), and range (Min, Max) were used as appropriate for descriptive statistics for summarizing the continuous data. Categorical data were presented as the number and percentage of patients in each category. Percentages were based on number of patients with data at a certain time point in the analysis set of interest except for the presentations of medical history, medications, and AEs, where the percentages were based on the number of patients in the study population. Both continuous and categorical data were tabulated by dose group and by scheduled time point in relation to the baseline, which was defined as the last nonmissing value before the first administration of SBE or the placebo. If there were several values at a scheduled visit for a patient, the last observed value within the time window of the scheduled visit was used in the analyses. In the case that a patient withdrew prior to the last planned visit in the study, the patient data were included in the analyses up to the time of discontinuation. Least Square Means analysis was performed to evaluate wound pain (VAS) after SBE versus placebo administration.
Ethics
The study was performed in accordance with the national regulations, the Declaration of Helsinki, and The International Council for Harmonisation (ICH E6 (R2))/Good Clinical Practice (GCP).
Graphics
Figures were created with BioRender.com
RESULTS
Study population
Thirty-six patients were screened for eligibility to participate in the study. A total of 28 patients received the study treatment (24 patients received SBE, and 4 patients received the placebo treatment). One patient in the BID multiple-dose (MAD) group discontinued the study due to noncompliance with study protocol on Day 5 after having been treated with a total of 8 (out of 10) doses of SBE. This patient could not come to the clinic on Day 5 to receive the final treatment due to a health condition unrelated to the SBE treatment.
The median age (min-max) of the patients enrolled to the study was 73 (46–83) years and 76 (74–78) years in the pooled SBE group and in the placebo treatment group, respectively. Fourteen patients were female, 13 of whom were randomized to the SBE treatment groups, and one patient was randomized to the placebo treatment group. The median weight (min, max) was 108.5 (63–198) kg and 85.5 (63–94) kg in the pooled SBE treatment group and in the placebo treatment group, respectively. A summary of the demographics of the patient participating in the study is presented in Supplementary Table S3. All prior medication reported in the study was ongoing, on and after the (first) administration of SBE or the placebo and was therefore reported as concomitant medication. As this was a safety study different types of chronic leg wounds were included (please see inclusion and exclusion criteria above); however, details on exact cause(s) for each wound were not collected. We do though know that our patient population at the wound center is mainly patients with venous leg ulcers (venous hypertension) of which approximately 25% also suffer from diabetes, and some have other contributing comorbidities like mobility impairment and obesity. Thereby most of the wounds were expected to have complex etiologies.
SBE dose escalation
Although there were no safety concerns identified, by the SMC, in the SAD phase of the study for any of the SBE formulations tested, the SMC decided to continue the treatment of the patients in the MAD phase of the study with the SBE formulation at 500 µg/mL HOCl + 2% HAc. The main reason was as follows: the results obtained with the 1000 µg/mL HOCl + 3% HAc formulation were similar to those obtained with the 500 µg/mL HOCl + 2% HAc formulation. Therefore, there was no added value of using a formulation at a concentration that was higher than necessary to achieve the same result.
Safety
No serious AEs (SAEs) were reported in any of the treatment groups. In total, five AEs in two patients treated in the MAD groups were registered (Supplementary Table S4). One patient in the OD group reported two AEs, one was in musculoskeletal and connective tissue disorders [Preferred Term (PT) myalgia] and one AE in nervous system disorders (PT syncope). One patient in the BID group reported three AEs, two were in gastrointestinal disorders (PT vomiting or PT nausea) and one was decreased hemoglobin (PT hemoglobin). None of the AEs was related to the SBE treatment, and four of them were resolved before the completing of the study.
For the laboratory parameters analyzed, a total of eight abnormal values of clinical significance were detected in three patients. Only one of the clinically significant abnormal values was developed postbaseline and corresponded to PT decreased hemoglobin for a patient in the BID group in the MAD phase of the study. No clinically significant abnormal values for physical examination, vital signs, or ECG were reported in any of the study groups.
Wound pain
The topically applied SBE was well tolerated at all concentrations tested. Overall, there were low values reported for leg wound pain in all study groups. Median values at baseline (i.e., Day 1, predose) varied from 0 to 10.5 mm on the 0–100 mm VAS. The maximum postbaseline VAS values for wound pain varied from 0.5 to 10.5 mm in the SAD groups with the highest median (min, max) value of 10.5 (0, 49) mm in the group treated with 1000 µg/mL HOCl + 3% HAc, this highest median was seen 2 min after SBE treatment, and the median stayed at 9.5–10 mm up until the 20-min evaluation and had dropped to 2 mm at the 40-min evaluation point post-treatment. In this group the median VAS after dressing removal was also 10.5 (0, 22), increased from 1 (0, 21) predressing removal. The same group reported also the highest maximum change (max value) from baseline in wound pain with a VAS median value of 1 (0, 27) mm 2 min after treatment. The corresponding values for maximum change from baseline in the other SAD groups were as follows: 0.5 (0, 21) mm in the group treated with 500 µg/mL HOCl + 1% HAc 60 min after treatment; 2 (−1, 46) mm in the group treated with 500 µg/mL HOCl + 2% HAc 15 min after treatment; and 1 (0, 37) mm in the group treated with 500 µg/mL HOCl + 3% 2 min after treatment. For the placebo group the maximum change from baseline was seen 15 min after treatment as a VAS median of 0 (−5, 16) mm.
The maximum median (min, max) change from baseline in wound pain was similar in both the OD and BID multiple-dose groups with VAS median values of 0 (−32, 67) mm and 10.5 (0, 45) mm, respectively. For both groups these highest median differences from baseline were seen 20 min after treatment, and had lowered after 40 min. The OD and BID groups were also similar in terms of maximum value and maximum change in wound pain from the last SBE dosing/treatment.
The statistical analysis of the change from baseline in wound pain comparing each of the SAD groups or the pooled SBE group with the placebo treatment group did not show any differences between the groups at any of the timepoints where wound pain was evaluated (2 min to 3 h after each treatment) (Supplementary Table S5).
Wound size
The wound size at baseline varied among the treatment groups with median (min, max) values ranging from 3.95 (1.8, 6) cm2 in the SAD group treated with 500 µg/mL HOCl + 3% HAc formulation to 29 (1.5, 50) cm2 in the BID MAD group treated with 500 µg/mL HOCl + 2% HAc formulation. The median (min, max) wound size at baseline was 6.0 (1.5, 50) cm2 in the pooled SBE treatment group and 4.23 (3.0, 7.2) cm2 in the placebo group. All wound size data are presented in Table 2.
Descriptive statistics of wound size/area (cm2)
Baseline is the last value before the (first) administration of SBE or the placebo.
SBE, SoftOx Biofilm Eradicator; HOCl, hypochlorous acid; HAc, acetic acid; SD, standard deviation; OD, once daily; BID, bis in die (Latin), twice daily.
The wound size was reduced over time in all the treatment groups irrespective of the SBE formulation evaluated (Table 2). The change from baseline in wound size measured at the follow-up visit was −1.22 (−17.95, 12.65) cm2 and 0.08 (−1.5, 1.5) cm2 for the pooled SBE treatment groups and the pooled placebo group, respectively. The corresponding wound size changes in the SAD groups were as follows: −2.18 (−4, −0.22) cm2 in the 500 HOCl µg/mL Cl + 1% HAc group, −0.16 (−3, 12.65) cm2 in the 500 µg/mL HOCl + 2% HAc group, −0.41 (−1.19, 0) cm2 in the 500 µg/mL HOCl + 3% HAc group, and −0.89 (−2.05, −0.48) cm2 in the 1000 µg/mL HOCl + 3% HAc group. A bigger absolute reduction in wound size was measured in the MAD groups, where the median wound size change was −2.99 (−14.25, −1.5) cm2 in the OD group and −10.48 (−17.95, −0.38) cm2 in the BID group.
Bacterial presence/bioburden
Bacterial burden in the wounds was reduced in all treatment groups, including the placebo treatment group, but the percentage reduction was numerically higher in the SBE treatment groups compared with the placebo group. The bacterial burden data in absolute numbers and percentages at day 1, day 5, and at follow-up are shown in Supplementary Table S6. The median (min, max) bacterial burden at baseline was similar in the pooled single-dose SBE treatment group with 1.5 × 105 (1.7 × 104, 5.2 × 106) CFU/mL and the placebo group with 2.2 × 105 (6.1 × 104, 3.5 × 106) CFU/mL. The median bacterial burden in the SBE treatment groups at baseline varied from 8.1 × 104 (2.4 × 104, 2.0 × 106) CFU/mL in the 500 µg/mL HOCl + 1% HAc SAD group to 1.1 × 106 (1.7 × 104, 5.2 × 106) CFU/mL in the 500 µg/mL HOCl + 2% HAc SAD group.
The median change in bacterial burden in the pooled single-dose SBE group after treatment was −98% (−100%, 476%), whereas in the pooled placebo group it was −49% (−40%, −70%). The median (min, max) percentage change in bacterial burden from baseline to the follow-up visit in the treatment groups was as follows: −94% (−100%, 138%) CFU/mL in the 500 µg/mL HOCl + 1% HAc group, −99% (−100%, −50%) CFU/mL in the 500 µg/mL HOCl + 2% HAc group, 92% (43%, 479%) CFU/mL in the 500 µg/mL HOCl + 3% HAc group, and −84% (−98%, 525%) CFU/mL in the 1000 µg/mL HOCl + 3% HAc group. In the pooled placebo group the median (min, max) percentage change was −19% (−94%, 479%). For the MAD groups, the median (min, max) percentage change in bacterial burden from baseline to the follow-up was −91% (−93%, −86%) CFU/mL in the OD group and −60% (−91%, 519%) CFU/mL in the BID group. Within the groups, the numerical reduction in bacterial burden from the predose to the follow-up visit was more consistent in the MAD groups with 7 out of 8 subjects showing a reduced bacterial burden compared with 10 out of 16 subjects in the SAD groups.
DISCUSSION
In the current clinical first-in-human study, the treatment with SBE, consisting of HOCl and HAc, was overall found to be safe and well tolerated in patients with chronic leg wounds. No SAEs were reported in the study, and only two patients treated with SBE in the MAD groups reported a total of five AEs that were not related to the treatment. There were no abnormal values of clinical significance detected for any physical examination, vital signs, and ECG measurements. Of the total of eight abnormal laboratory parameters that were of clinical significance, only one value developed at the follow-up visit, i.e., was not identified either at the screening or at the first treatment day (baseline). This value was linked to a patient in the BID MAD group and corresponded to levels of hemoglobin that were below the lower limit of the normal reference range.
Pain originating from leg wounds is common following debridement or irrigation of wounds, 41 and it was therefore selected as the main tolerability parameter for the topical application of SBE in the current study. The treatment of leg wounds with SBE at all concentrations tested was not associated with increased perception of pain compared with placebo. The statistical analysis of the change from baseline in wound pain comparing each of the SAD groups or the pooled SBE treatment group with the placebo group did not show any differences between the groups at any of the timepoints where pain was evaluated. Overall, there were numerically higher values for pain reported in the MAD groups compared with the SAD groups. However, there was no difference observed between the OD and BID treatment groups, indicating that the higher pain score could be due to the repeated irrigation of the wounds and not the treatment of the wounds with SBE. Other HOCl containing wound irrigation products on the market have also proven safe and tolerable to patients; however, the difference for SBE is the combination of HOCl and HAc, which is expected to make the drug product, SBE, more potent in healing wounds and killing bioburden. This should be tested in forthcoming phase 2 and 3 trials. Furthermore, in this combination product HAc stabilizes HOCl and thereby improves the stability of HOCl, which is a rather volatile molecule.
Within the SBE treatment groups, the numerical reduction in bacterial burden from predose to the follow-up visit evaluation was more consistent in the MAD groups compared with the SAD groups. The data did not show dose-dependent differences among the SAD groups, but for the MAD groups, a better efficacy trend was seen in the BID group compared with the OD group. However, it is important to note that this study was not dimensioned to show efficacy or dose response of the SBE treatment.
Wound closure or wound size also seemed positively affected by the SBE treatment compared with the placebo treatment. A reduction in wound size from the baseline values was observed over time in all SBE treatment groups irrespective of the SBE formulation evaluated. The median change in wound size from the baseline value to the follow-up visit value was −1.22 cm2 in the pooled SBE treatment groups, whereas the wound size increased 0.08 cm2 in the pooled placebo group. A bigger absolute reduction in wound size was observed in the MAD groups, where the median change from the baseline value to the follow-up visit value was −2.99 cm2 in the OD group and −10.48 cm2 in the BID group. Considering that the BID group daily received the double dose used in the OD group, it is tempting to conclude that a trend of dose-dependent reduction in wound size was observed in the MAD groups treated with SBE. As HOCl and HAc react immediately with the wound, including bacteria, and have no systemic uptake or prolonged release, the action will only occur during the 20-min treatment, which might explain the better efficacy of the BID treatment compared with OD treatment in relation to antibacterial efficacy and wound size reduction. Again, as these were secondary endpoints the trial was not designed and powered to statistically support such findings. The detected reductions in wound size were due to epithelialization, and improved epithelialization has also been proved for another similar HOCl and HAc solution. 42
Limitations to the study need to be noted. Due to the exploratory nature of the study, no formal sample size calculation was performed, and the sample size was limited to 24 patients receiving SBE treatment (in six groups). The SAD groups contained only one patient treated with the placebo each and the MAD groups contained none, which prevented us from directly attributing the positive effects seen on wound size and bacterial burden reduction to the SBE treatment. Furthermore, complete wound closure is a preferred endpoint in a clinical study. However, wound healing is a lengthy process; hence, complete wound closure as an endpoint requires longer duration of follow-up of the patients. In the current study, the follow-up period was limited to 2–5 days, as safety was the main endpoint, and therefore, wound size reduction was used as a surrogate exploratory endpoint. Any future study investigating the SBE treatment of wounds should have a longer follow-up period, e.g., three months.
In summary, this first-in-human clinical study demonstrated that SBE applied on chronic leg wounds was safe and well-tolerated. There were no statistically significant differences in the evaluation of pain during and after the wound treatment procedure comparing SBE with the placebo. For the secondary endpoints, the treatment with all SBE formulations reduced the number of bacteria in the wounds. A dose-dependent reduction in wound size was observed in the MAD SBE treatment groups.
In conclusion, the results from this study support the positive safety and tolerability profile of SBE treatment. The results also indicate that the multiple-dosing regimen would be preferable to a single-dose regimen in terms of potential efficacy on bacterial burden and wound size for further evaluation in future clinical trials.
KEY FINDINGS
Positive safety and tolerability profile of SBE treatment on chronic leg ulcers
Antimicrobial effect on chronic leg wounds after SBE treatment
Decreased wound size after SBE treatment of chronic leg ulcer patients
Footnotes
ACKNOWLEDGMENTS AND FUNDING SOURCES
The authors are grateful to all the patients who participated in the study and the excellent study nurses: Stine Ingvartsen, Lisbeth Vorbeck, and Stine Ørts Pedersen. The authors also thank Lasse Andersen Kvich at Costerton Biofilm Center, University of Copenhagen, for the microbiological analyses in the clinical study.
The study was sponsored by SoftOx Solutions AS, Fornebu, Norway, with partial funding from the United States Department of Defense.
AUTHOR DISCLOSURE AND GHOSTWRITING AND ABOUT THE AUTHORS
D.P.S. was the principal investigator at the study site. T.B. is the manager of the study site. G.G., K.K.M., and T.B.J. designed the study. M.M.F. wrote the article, and E.J. critically reviewed the data and the article. All authors approved the article.
SUPPLEMENTARY MATERIAL
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
List of Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.