
Abstract
Abstract
Wnt signaling is a major player during development and its misregulation often leads to disease, especially cancer. The negative feedback Wnt regulator homologs, Nkd1 and Nkd2, have been shown to inhibit Wnt signaling during development, and current evidence suggests that Nkds degrade Dvl proteins to antagonize Wnt signaling. Here, we demonstrate that during early zebrafish development Nkd1 does not alter either endogenous or exogenous levels of Dvl2. Furthermore, Dvl2 does not affect the levels of Nkd1. Cumulatively, these results demonstrate that Dvl2 is a ubiquitous and stable protein and that Nkds may not always function to degrade Dvl proteins as a method of inhibiting Wnt signaling.
Introduction
As Nkd and Nkd1 have been shown to bind other Dvl proteins,4,8,10 we wanted to further investigate the role of Nkd1 in the stability of Dvl2. To investigate the interaction between Nkd1 and Dvl2, we used the early zebrafish embryo for which the Wnt signaling pathways and timing of Wnt signaling are well characterized.11–13 To investigate endogenous Dvl2, a polyclonal antibody to zebrafish Dvl2 was generated. In contrast to recent findings,1–3 this study demonstrates that during early zebrafish development Dvl2 is a stable protein and is not involved in hastening the degradation of Nkd1. Nkd1 also does not increase the degradation rate of either endogenous or exogenous Dvl2.
Results
Characterization of anti-Dvl2 antibody
To investigate the role of endogenous Dvl2, a polyclonal antibody to zebrafish Dvl2 was generated. The epitope was designed in a nonconserved region of Dvl2 and is not predicted to cross react with Dvl1 or Dvl3 (Fig. 1A), although this has not been formally tested. Affinity-purified polyclonal anti-Dvl2 can detect both exogenous and endogenous Dvl2 proteins when used as low as 1:20,000 for western analysis (Fig. 1B). Knockdown of endogenous Dvl2 with translation blocking antisense oligonucleotide morpholinos (MO) was observed with the antibody, confirming that the antibody can detect endogenous Dvl2 (Fig. 1C, D). To determine if the antibody could detect exogenous Dvl2 by immunohistochemistry, Dvl2HA was injected into one of four blastomeres to generate mosaic Dvl2HA expression. The antibody could detect Dvl2HA at a dilution of 1:5000 with similar, but weaker staining in non-Dvl2HA-expressing cells (Fig. 1F, I). The HA and the Dvl2 antibodies give nearly identical staining patterns and detect cellular puncta, which is characteristic of Dvl staining and is very similar to Drosophila Dsh and mammalian Dvl2 proteins.14–17 At a higher concentration (1:1000), endogenous Dvl2 staining is very similar to that seen at 1:5000 in both Dvl2HA-expressing and -nonexpressing cells; only the staining is more robust and is ubiquitous (Fig. 1G). In a co-labeling experiment, there is substantial overlap between the HA and the Dvl2 antibodies (Fig. 1H–J). We conclude that the antibody to zebrafish Dvl2 is robust and can detect endogenous Dvl2 by both western analysis and immunohistochemistry.

Characterization of zebrafish anti-Dvl2 antibody.
Dvl2 is stable, but Nkd1 is not
Previously, it has been demonstrated in cell culture that Dvl proteins are targeted for degradation via several mechanisms.1,3,10,16,18,19 Therefore, we wanted to determine the stability of endogenous Dvl2 during zebrafish development as well as in the presence of Nkd1. First, endogenous Dvl2 protein levels were assayed over a 3-h period in the presence of cycloheximide (chx), a protein synthesis inhibitor starting at dome stage (4.3 hours postfertilization [hpf]). Over this period, Dvl2 protein levels did not diminish to any noticeable degree (Fig. 2A). A 4–5-h exposure of zebrafish blastula to chx was lethal, so longer assays were not feasible. The stability of Dvl2 is further emphasized by the MO knockdown studies. Within the first 18 h of development, increasing concentrations of a translation blocking Dvl2 MO had no effect on the levels of Dvl2, either by western analysis or by immunohistochemistry (not shown). Only after 24 hpf could a dose-dependent decrease in Dvl2 proteins be observed (Fig. 1C, D). As this Dvl2 MO blocks protein translation, this suggests that Dvl2 proteins are very stable in the early zebrafish embryo.
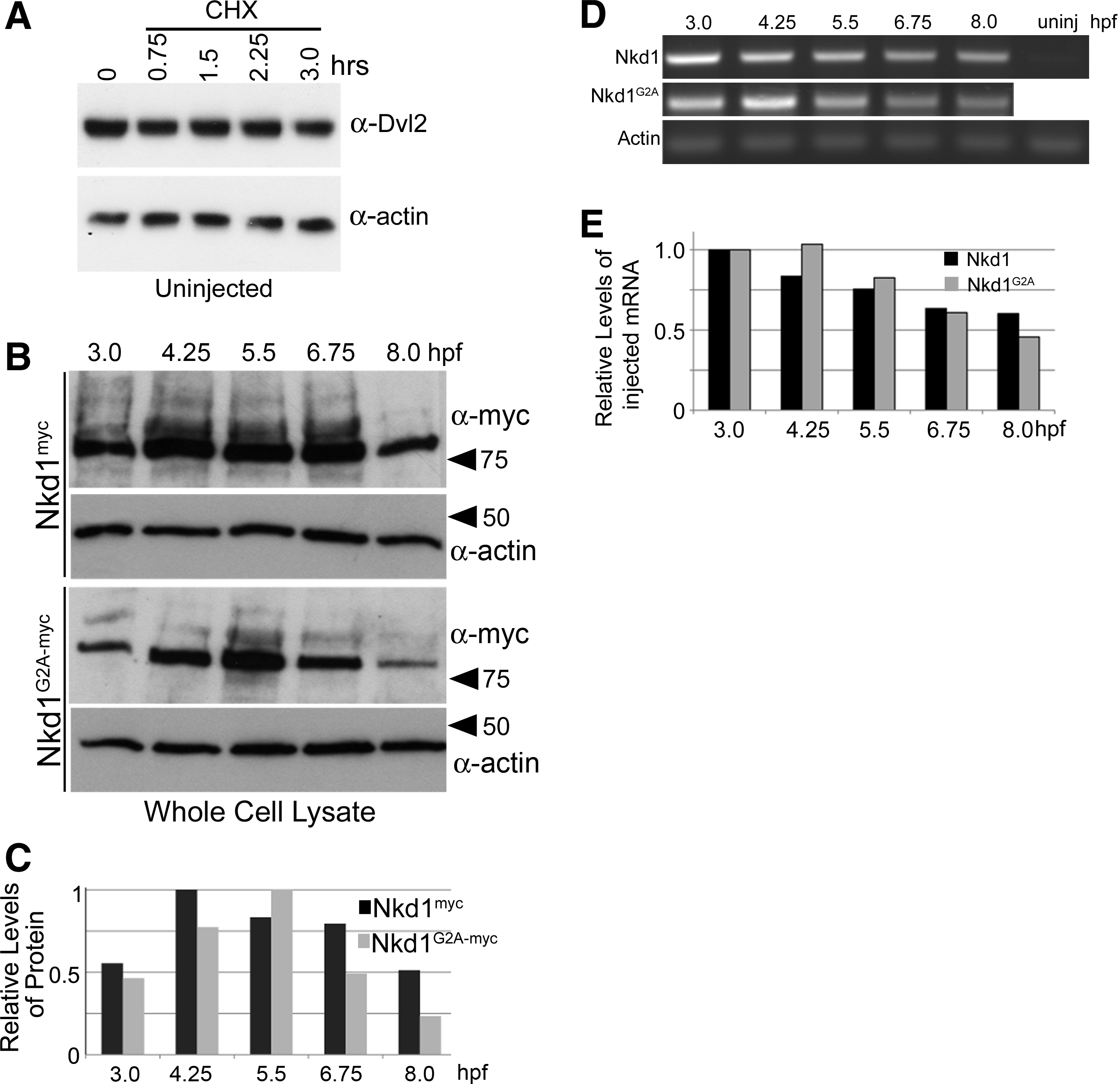
Dvl2, but not Nkd1, is a stable protein.
To determine the effect of Nkd1 on Dvl2 protein levels, we first wanted to determine the changes in Nkd1myc protein levels without chx treatment over time. As expected, the levels of protein increased with continued mRNA translation, but after 6.75 hpf, levels started to decrease (Fig. 2B). The ability of Nkd1 to effectively inhibit Wnt signaling is dependent on an N-terminal myristoylation sequence.5,9 Mutating the second glycine to alanine (G2A) dramatically reduces the ability of Nkd1 to inhibit Wnt signaling. 9 Similar to Nkd1myc, the levels of Nkd1G2A-myc protein levels decrease over time, but in a manner that appears different than Nkd1myc (Fig. 2B, C). This experiment has been repeated with different time points and different epitopes with similar conclusions. This apparent difference in stability between Nkd1myc and Nkd1G2A-myc cannot be attributed to differential mRNA degradation, as the injected mRNA levels were similar between Nkd1myc and Nkd1G2A-myc over time as detected by reverse transcription (RT)-polymerase chain reaction (PCR) (Fig. 2D, E).
Nkd1 does not destablize Dvl2
Nkd proteins are predicted to act at the level of Dvl and one hypothesis is that Nkds destabilize Dvl, preventing further Wnt signaling.1,2 To test this hypothesis and determine if Nkd1 affects Dvl2 protein stability, Nkd1myc mRNA was injected into embryos and the levels of Nkd1myc and endogenous Dvl2 were assayed in the presence of chx. A near-linear decrease in Nkd1myc protein levels (half life = 1.8 h) over 3 h of chx treatment was observed, whereas no changes were observed in endogenous Dvl2 protein levels over the same time period (Fig. 3A). It is possible that Nkd1myc only affects a small pool of Dvl2 that is undetectable by our methods. Such phenomena have been described previously.10,17,20 To determine if Nkd1 altered exogenous Dvl2 protein levels, or if exogenous Dvl2 altered Nkd1 protein levels, we co-injected Nkd1myc and Dvl2HA mRNAs and assayed for protein levels over a 3-h period in the presence of chx. Similar to endogenous Dvl2, no significant difference in the degradation of Dvl2HA in the presence or absence of Nkd1myc was observed (Fig. 3B–D). Furthermore, excess Dvl2HA did not significantly increase the degradation rate of Nkd1myc (half life = 1.70; Fig. 3C, E). To determine if there is an effect at later embryonic stages, we performed a similar assay at 80% epiboly (but without chx treatment) when there is significant Wnt signaling patterning the anterior-posterior neuroectoderm.11,13 We harvested embryos from the same set of injections at 30% and again at 80% epiboly and compared the levels of Dvl2HA with and without Nkd1myc at these two time points (Fig. 3F, G). At 80% epiboly we observed an approximately 50% decrease in the levels of Dvl2HA, in both the Dvl2HA injected alone or Dvl2HA co-injected with Nkd1myc (Fig. 3F, G). We also did not observe any changes in the levels of endogenous Dvl2 (Fig. 3F). Thus, we conclude that Nkd1 does not destabilize Dvl2 at several stages of early zebrafish development.
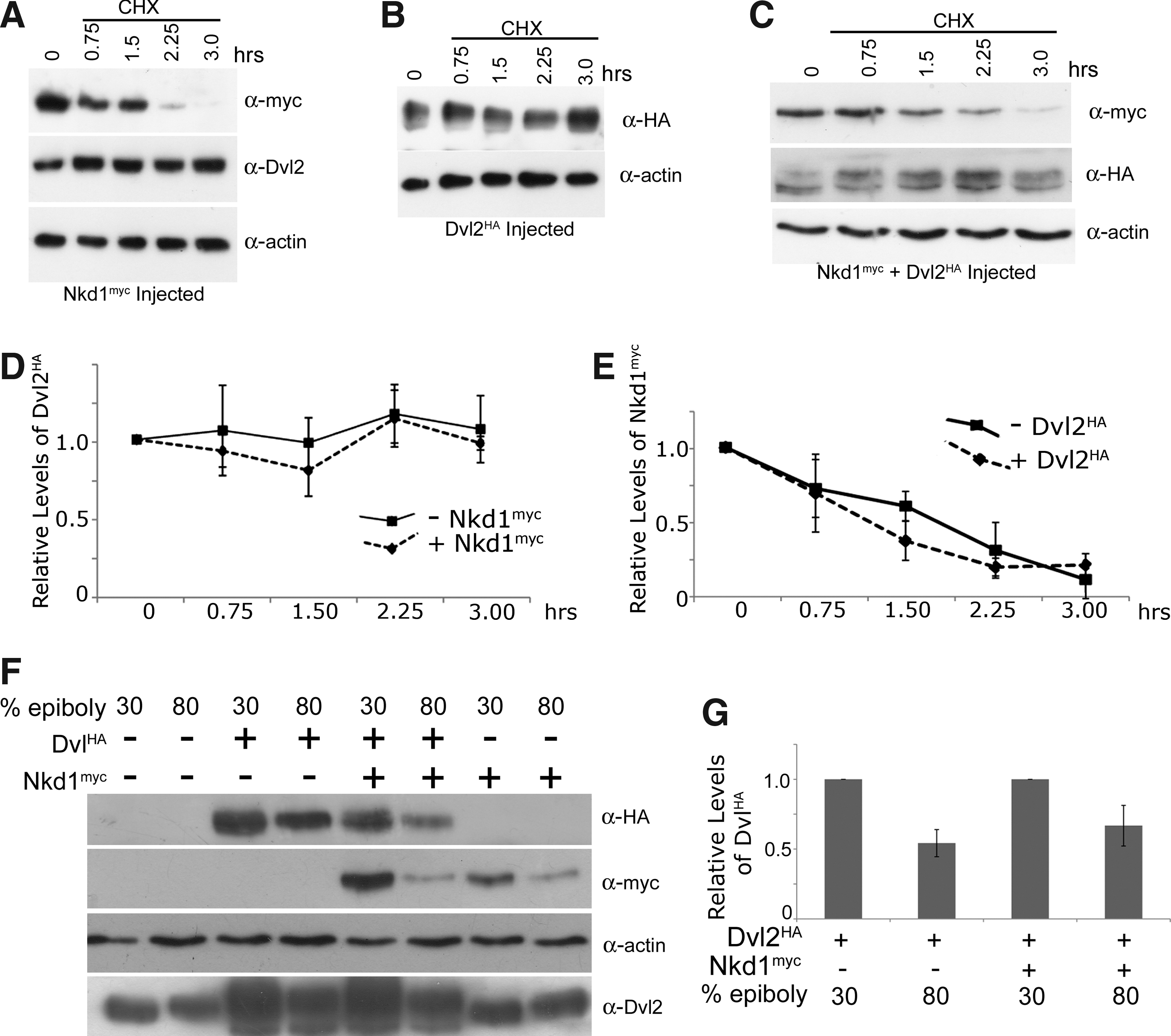
Nkd1 does not alter Dvl2 stability. At 4.3 hpf (t = 0), Nkd1myc
In a previous study it was demonstrated that an intact myristoylation sequence was required for the ability of Nkd2 to degrade Dvl1 and that Nkd2G2A was more stable than Nkd2 wild type. 3 In zebrafish, Nkd2G2A is also at least as stable as wild-type Nkd2 (unpublished). However, evidence with Nkd1G2A in zebrafish suggests the opposite: Nkd1G2A is less stable than Nkd1 (Fig. 2B, C). This was confirmed in the chx experiments, which demonstrated that, in contrast to human and zebrafish Nkd2, zebrafish Nkd1G2A is much less stable than wild-type Nkd1 with only approximately 10%–15% of the protein remaining after 1 h of chx treatment (Fig. 4A, B). This experiment has been repeated with different time points and different epitopes with similar results and in fact it is often difficult to detect any Nkd1G2A protein after 1 h of chx exposure. Exogenous Dvl2HA had no effect on the stability of Nkd1G2A-GFP, nor was there any effect of Nkd1G2A-GFP on the stability of Dvl2HA or endogenous Dvl2 (Fig. 4B, C and not shown). However, with such a rapid rate of Nkd1G2A decay, it is not possible to determine if Nkd1G2A has any real effect on Dvl2HA or endogenous Dvl2 stability. The rapid rate of Nkd1G2A-GFP decay suggests that either membrane association of Nkd1 confers stability or converting the second amino acid glycine to alanine makes proteins like Nkd1G2A highly unstable. 21
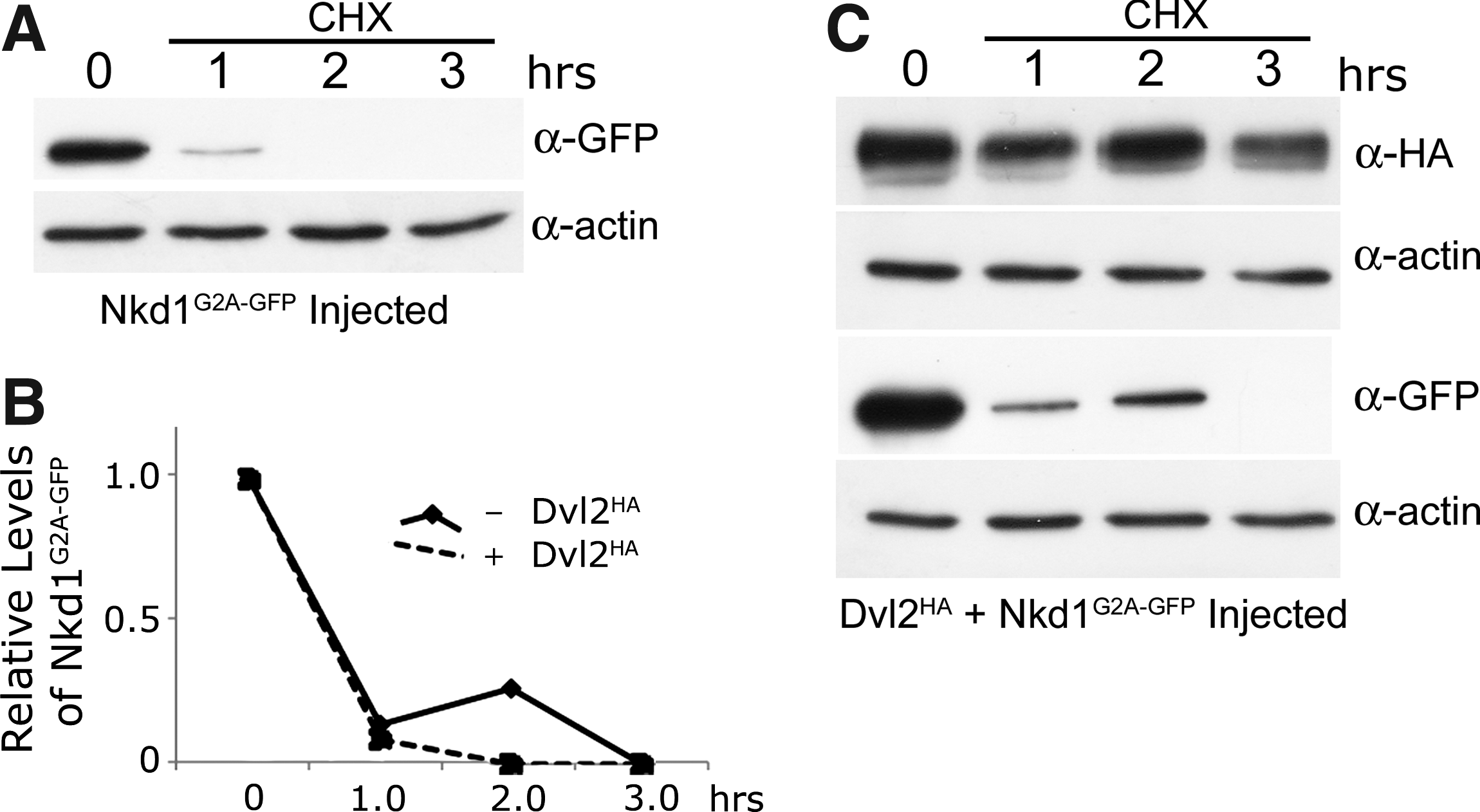
Nkd1G2A-GFP is very unstable.
Nkd1 degradation is mediated by poly-ubiquitin-mediated proteosome degradation
To determine if Nkd1 is degraded by ubiquitin-mediated proteosome degradation, Nkd1myc or Nkd1G2A-myc mRNA, together with Ub-HA mRNA, was injected into zebrafish embryos and immunoprecipitated with anti-myc and western blotted for anti-HA. These experiments were carried out in the presence or absence of a proteosome inhibitor cocktail. These results demonstrated that wild-type Nkd1 was efficiently labeled with ubiquitin, the levels of which increased in the presence of proteosome inhibitors (Fig. 5). Consistent with the chx treatments, this suggests that Nkd1 is efficiently turned over by ubiquitin-mediated proteolysis. In contrast, the levels of poly-ubiquitin-labeled Nkd1G2A-myc were substantially lower despite its apparently more rapid rate of decay when compared to wild-type Nkd1 (Fig. 5). Also, when the western blot was probed with anti-myc, the levels of Nkd1myc and Nkd1G2A-myc are similar in the presence of proteosome inhibitors and the levels of Nkd1G2A increased in the presence of the proteosome inhibitors (Fig. 5 IP: α-myc; WB: α-myc lanes 2 and 4). This suggests that Nkd1G2A degradation is mediated via the proteosome, but why there is less Ub-HA staining relative to wild-type Nkd1 is unclear, when in fact, higher levels of Nkd1G2A labeled with Ub-HA were expected. One possibility is that Nkd1G2A is mono-ubiquitinated, the levels of which could not be detected on this western blot.

Nkd1 is ubiquitinated. Nkd1myc or Nkd1G2A-myc mRNAs were co-injected with 200pg of UbHA mRNA, and at 3.8 hpf (sphere stage) half of the embryos were treated with proteosome inhibitors (P.I.) for 2 h with the other half being controls. At 5.8 hpf cells were lysed and proteins were immunoprecipitated with anti-myc antibodies, western blotted, and probed with anti-HA or anti-myc (top two panels). Before incubation with anti-myc, a sample of whole cell lysate was removed for analysis with anti-HA, anti-myc, and anti-actin (bottom set of panels).
Discussion
The effect of Nkd1 on Dvl2 protein levels in the early zebrafish contrasts with recent findings on the effect of Nkd2 and Dvl1 in culture, where it was found that Nkd2 and Dvl1 form a mutual destruction complex and enhance each other's degradation. 3 Also, Nkd1 was found to regulate Dvl1 levels through the PP2A regulatory β-subunit PR72. 1 This suggests that Nkds may destabilize Dvl1, but the current study suggests that Nkd1 does not affect Dvl2 levels during early zebrafish development. This observation is likely due to differences in Dvl stability in these different models. In the aforementioned studies, there are changes in endogenous Dvl levels as observed by siRNA experiments, either targeting Dvl1 itself or Nkd1 and observing the effect on endogenous Dvl levels.1,3 We also observed an effect of Dvl2 knockdown at 1 day postfertilization (dpf). Thus, it is likely that highly stable Dvl2 protein is a phenomenon of early zebrafish development, but the significance of this is unknown.
The short half-life of Nkd1 (1.8 h) and the turnover of the injected mRNA suggest that there is insufficient Nkd1 protein levels to affect later aspects of Wnt signaling during development. This is consistent with the lack of an Nkd1 overexpression phenotype at 1 dpf. 5 Nonetheless, Nkd1 is both necessary and sufficient to inhibit both endogenous and exogenous Wnt signaling at stages that are consistent with the current study,5,9 despite the lack of Dvl2 degradation. This argues that Dvl2 degradation is not a prerequisite for Nkd1 antagonism of Wnt signaling.
This study also contrasts with a recent finding in zebrafish that suggests that Nkd1 can induce the degradation of Dvl2. 2 The discrepancy between these two studies is unclear, but may be related to experimental design. In our experiments we observed that exogenous Dvl2HA protein did decrease between 30% and 80% epiboly, but that exogenous Nkd1myc did not enhance the rate of Dvl2HA degradation.
In summary, this study demonstrates that during early zebrafish development, Dvl2 is a ubiquitous and stable protein, which is not degraded in the presence of Nkd1. Furthermore, the half-life of Nkd1 is approximately 1.8 h, which is not significantly altered by Dvl2, but is significantly altered by the presence of an intact myristoylation sequence. This suggests that the function of Nkd proteins is either critically dependent on the environment or that Nkd1 and Nkd2 have fundamentally different properties when it comes to antagonizing Wnt signaling. Thus, at least during early zebrafish development, Nkd1 does not antagonize Wnt signaling by degrading Dvl2 proteins.
Materials and Methods
Fish maintenance
Adult zebrafish and embryos were raised at 28.5°C and were staged by anatomical criteria or hours postfertilization according to Kimmel et al. 22 All experiments were performed in accordance with local, provincial, and federal guidelines and regulations (Animal Use Protocol no. 08R109).
Constructs
For pCS2+Uβ−HA the tandem Uβ−HA (8×) fragment was isolated from pMT123 (kindly provided by Dr. Michael Freeman, Vanderbilt University) by an NotI-EcoRI double digest, blunt ended and ligated into pCS2+ digested with StuI. pCS2+Nkd1myc has been previously described 5 and pCS2+Nkd1GFP was constructed by replacing the 6× myc tags in Nkd1myc with GFP. pCS2+Nkd1G2A-myc or pCS2+Nkd1G2A-GFP was created by site-directed mutagenesis on the parent clones with the following primers Nkd1G2A-For: 5′-GCAGGATCCCATCGATGATGGCTAAACTTCATTCC-3′; Nkd1G2A-Rev: 5′-GGAATGAAGTTTAGCCATCATCGATGGGATCCTGC-3′ following standard procedures. Dvl2 ATG Morpholino: 5′-TAAATTATCTTGGTCTCCGCCATGT-3′. pCS2+Dvl2HA was kindly provided by Lila Solnica-Krezel.
mRNA injections
Capped mRNA was synthesized using Ambion's mMessage mMachine kit. After transcription, the RNA was purified over a G-50 sephadex column (Roche) or with the Megaclear kit (Ambion) and diluted in RNase free water, and its quantity and quality was analyzed as described above. The RNA was pressure injected at a final concentration of 800 ng/μL nkd1myc or nkd1GFP; 1200 ng/μL of nkd1G2A-myc or nkd1G2A-GFP; 200 ng/μL Uβ−HA in 1% phenol red into the yolk cell of one or two-cell stage embryos.
RT-polymerase chain reaction
At least 15 embryos from each time point were snap-frozen in liquid nitrogen in the presence of 500 μL of Trizol. Total RNA was extracted using manufacturers guidelines and converted to cDNA using Superscript III (Invitrogen). PCR primers to Nkd1 (5′-GAAGCGCTGCACTACAGG-3′) and the T7 promoter of pCS2+ (5′- TAATACGACTCACTATAG-3′) were used for both exogenous Nkd1- and Nkd1G2A-injected embryos. Actin (For: 5′- ATGCCCCTCGTGCTGTTTT-3′; Rev: 5′-TCTGTCCCATGCCAACCAT-3′) was used as a loading control. Standard PCR conditions were carried out over 30 cycles and loaded onto a 1% gel for electrophoresis.
Immunohistochemistry
Injected embryos were fixed at 30% epiboly in 4% PFA overnight at 4°C, washed four times in 1× phosphate-buffered saline (PBS) with 0.1% Tween20, incubated for 1 h at RT in block (5% goat serum, 5 mg/mL bovine serum albumin, 2% dimethyl sulfoxide [DMSO], 0.1% Tween20 in 1× PBS), and incubated with primary antibody in block overnight at 4°C. After primary incubation, embryos were washed 6× in 1× PBS with 0.1% Tween20 and 2% DMSO, incubated in secondary in block for 3 h at RT, washed 6× in 1× PBS with 0.1% Tween20 and 2% DMSO, put into 50% glycerol/PBS, deyolked, and mounted on slide with raised coverslip.
Primary antibodies used were Rat anti-HA 1:200 (Roche), and Rabbit anti-Dvl2 1:1000 or 1:5000. Secondary antibodies used were Cy2 Donkey anti-Rat 1:200 (Invitrogen); Cy3 Goat anti-Rabbit 1:200 (Invitrogen); Cy2 Goat anti-Rabbit 1:200 (Invitrogen); and syto59 1:10,000 (Invitrogen).
Cycloheximide treatments
Approximately 50–60 injected and well-developing embryos were dechorionated at sphere stage (3.8 hpf) in 0.3× Danieau. About 50 μM Chx (BioShop Canada) in 2% DMSO, 0.3× Danieau was preheated to 28.5°C. At dome stage (4.3 hpf) embryos were placed in preheated Chx solution (except for 10 embryos, which were used for t = 0) and incubated at 28.5°C. Every 45 or 60 min, 10 embryos were removed and deyolked at room temp in 0.3× Danieau, put into 100 μL of RIPA buffer containing protease inhibitors, triturated 10 times with P100 pipettor, and stored on ice for 1 h, after which an equal volume of 2 × dithiothreitol (DTT) sample buffer was added. Samples were boiled for 5 min, centrifuged at 16,000 g for 1 min, and stored at −20°C for western analysis. Approximately 20 μL of lysate (equivalent of one embryo) was loaded on an 8% acrylamide gel.
Proteosome inhibitors and immunoprecipitations
Approximately 60 embryos were injected at the 1-cell stage with nkd1myc or nkd1G2A-myc RNA along with ubHA RNA. Individual proteosome inhibitors were dissolved in 100% DMSO. Cocktail inhibitors were diluted in 10% DMSO and 0.3 × Danieau to their final concentrations: 300 μM MG132, 300 μM ALLN, 2 μM Epoxomycin, and 10 μm Lactacystin (Calbiochem). Control embryos were placed in 10% DMSO, 0.3 × Danieau only. Embryos were put into cocktail inhibitor or control media at dome stage (4.3 hpf) and allowed to develop to Shield stage (6 hpf). At this stage, embryos were neither dechorionated nor deyolked. Egg water was removed and 500 μL of ice-cold modified Rubenfelds buffer (20 mM Tris, pH 8.0; 0.3% NP-40; 140 mM NaCl; 10% glycerol; 1 mM ethylene glycol tetraacetic acid [EGTA]; 15 mM MgCl2; 1 mM NaVanadate) with Complete Protease Inhibitior® (Roche) was added. Approximately 10–15 pulses with a power pestle was used to disrupt chorions and cell membranes. To further disrupt membranes, lysates were triturated with a 1.5-inch 18-gauge syringe 8 to 10 times. Another 500 μL of Rubenfelds buffer containing Complete Protease Inhibitor was added to lysate. Intact nuclei, large cell membrane fragments and chorion were pelleted by centrifugation at 960 g for 5 min at 4°C. The top 900 μL was moved to a fresh 1.5 mL tube. Two hundred microliters of modified Rubenfelds buffer containing Complete Protease Inhibitor was added to obtain a final concentration of one embryo/20 μL. Aliquots of 350 μL were placed into three tubes (equivalent of 17.5 embyros). The remaining 50 μL was used for input analysis. Then, 4 μL of anti-myc (Vanderbilt Antibody Core) antibody was added to one tube and incubated at 4°C overnight with gentle agitation. The following day, 25 μL of washed Sepharose Protein G (GE Healthcare) beads were added to the precipitates and incubated for another 4 h at 4°C. The Protein G mixture was centrifuged at 960 g for 30 s and the beads were washed 3 × with IP Wash Buffer (20 mM Tris, pH 8.0; 150 mM NaCl; 0.3% NP-40). Pellets were resuspended in 25 μL of 2× DTT sample buffer (100 mM Tris-HCl pH 6.8; 200 mM DTT; 4% sodium dodecyl sulfate; 20% gyclerol; 0.2% Bromophenol blue), boiled for 5 min, centrifuged at 16,000 g for 1 min, and either stored at −20°C or 20 μL was loaded for Western analysis.
Western analysis
Protein samples were loaded onto denaturing polyacrylamide sodium dodecyl sulfate gels, blotted onto polyvinylidene fluoride (PVDF), and probed with the following antibodies: monoclonal anti-β-actin (Sigma) 1:2000; Polyclonal anti-Dvl2 (1:5000); monoclonal anti-myc (Vanderbilt Antibody Core) 1:2000; polyclonal anti-GFP (Torrey Pines) 1:1000; mouse monoclonal anti-HA (Vanderbilt Antibody Core) 1:1000. To quantify the levels of protein, peak intensity measurements (Image J) of Dvl2, HA, Myc, GFP, and Actin bands were taken. Relative levels of protein were first normalized to actin and then compared to untreated, or the first time point, which was arbitrarily set to 1.
Footnotes
Acknowledgments
T.V.R. would especially like to thank Drs. Lila Solnica-Krezel and Robert Coffey for the opportunity to initiate this work in their respective labs. We would also like to thank the volunteer and student members of the lab for their help with this project. We would also like to thank Dr. Richard Mosser for critical reading of this article. This work was supported by a Canadian Foundation for Innovation grant to T.V.R.
Disclosure Statement
No competing financial interests exist.