
Abstract
Abstract
Retinitis pigmentosa (RP) affects 1/4000 individuals in most populations, and X-linked RP (XLRP) is one of the most severe forms of human retinal degeneration. Mutations in both the retinitis pigmentosa GTPase regulator (RPGR) gene and retinitis pigmentosa 2 (RP2) gene account for almost all cases of XLRP. The functional roles of both RPGR and RP2 in the pathogenesis of XLRP are unclear. Due to the surprisingly high degree of functional conservation between human genes and their zebrafish orthologues, the zebrafish has become an important model for human retinal disorders. In this brief review, we summarize the functional characterization of XLRP-causing genes, RPGR and RP2, in zebrafish, and highlight recent studies that provide insight into the cellular functions of both genes. This will not only shed light on disease mechanisms in XLRP but will also provide a solid platform to test RP-causing mutants before proposing XLRP gene therapy trials.
Introduction
Research with zebrafish (Danio rerio) has provided fundamental insight into the development of vertebrate animals based on its rapid and external development, coupled with optical clarity during embryogenesis. The zebrafish genome has been nearly completely sequenced, and many of the genes and pathways are conserved with those of humans. The zebrafish is increasingly used as a model to study a range of human diseases, including cardiovascular disease, cancer, neurodegenerative disease, and visual disorders. 6 To develop an understanding of disease processes, many zebrafish models of monogenic human genetic disease have been generated through forward-genetic screens. Following the forward-genetic screen, reverse genetic approaches, such as morpholino-mediated knockdown, are used to facilitate functional assays of genes of interest. 6 Morpholinos are molecules used in reverse genetic research to knockdown gene function that is to prevent the synthesis of a targeted protein. This is a powerful method to demonstrate the function of a whole protein or, by causing a specific exon to be spliced out of a protein. The method can help to determine the function of the region of a protein encoded by that exon.
The zebrafish neural retina shows strong evolutionary conservation, its anatomy, histology, and function closely resemble that of the human retina (Fig. 1). Zebrafish retinal neurogenesis starts very early, following an inner-to-outer retinal order typical of other vertebrate species. Retinal ganglion cells (RGC) are differentiated and becoming postmitotic between 28 h post-fertilization (hpf) and 32 hpf.7,8 The differentiation of RGC spreads rapidly into the temporal retina between 36 to 40 hpf. 9 Cells of the inner nuclear layer appear at 38 hpf and are postmitotic by 48 hpf.8,9 The photoreceptor cells start to differentiate at 50–54 hpf, the first outer segments appear at 54–60 hpf. Retina lamination begins at 32 hpf. Lamination has spread across most of the retina by 48 hpf and is fully established by 60 hpf. 9 Similar to the human retina, the zebrafish retina possesses seven major cell types, including six types of neurons (rod and cone photoreceptor, horizontal cells, bipolar cells, amacrine cells, and ganglion cells) and a single type of glial cells (Müller glia). 10 Unlike mouse and rat models, zebrafish cone photoreceptors are subdivided into four distinct classes sensitive to ultraviolet, red, green, and blue wavelengths. 11 Within the zebrafish retinal outer nuclear layer, cone cells of different subtypes are arranged in a highly ordered crystalline mosaic pattern. 12 Zebrafish have a cone-rich retina similar to the human retina, providing an excellent model for studying human retinal degeneration.
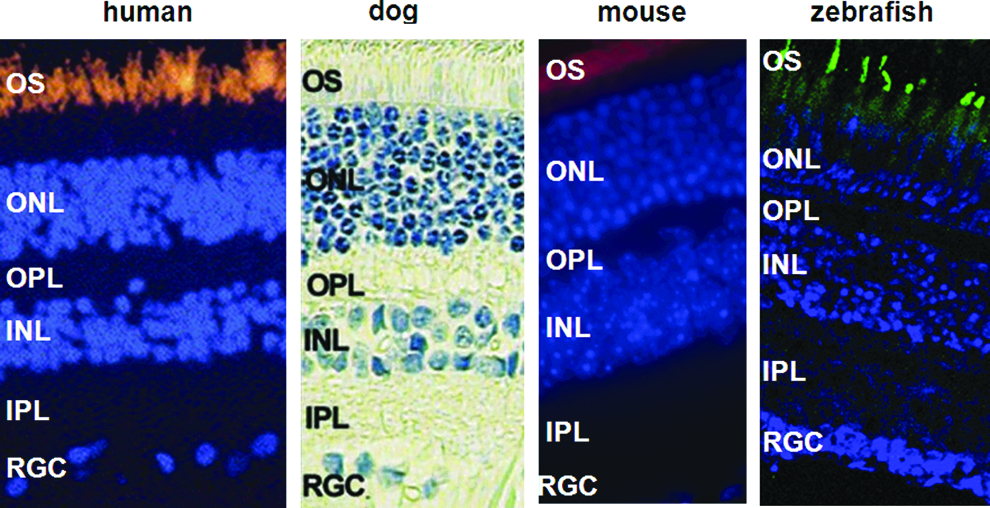
Similarities of retinal structure in human, dog, mouse, and zebrafish, which have been used to study RPGR function. Photoreceptor cell outer segments (OS) were labeled with anti-rhodopsin antibodies in human (orange), mouse (red), and zebrafish (green). INL: inner nuclear layer, IPL: inner plexiform layer, ONL: outer nuclear layer, OPL: outer plexiform layer, OS: photoreceptor cell outer segments, RGL: retinal ganglion cell layer. Photos of human retina and dog retina are modified from Adamian et al. 38 and Jiang et al., 62 respectively. Color images are available online at www.liebertpub.com/zeb
Early forward genetic screens for photoreceptor dystrophies have identified numerous mutations, that serve as a model for understanding the disease mechanisms of human photoreceptor degeneration.13,14 For example, the oval mutant zebrafish exhibits photoreceptor degeneration and kidney cysts. 13 Position cloning identified a nt779T>A mutation (L260X) in the gene encoding a component of the intraflagellar transport particles (ift88), suggesting that IFT88 has a role in the maintenance of the photoreceptor outer segment. 15 IFT particles are involved in the transport of proteins from the photoreceptor cell body to the outer segment along the connecting cilium. Mutations in zebrafish ift88, ift57, and ift172 genes resulted in opsin mislocalization and dishevelled cone cells. 16
Recently, reverse genetics has been used to characterize different human retinal disease genes in zebrafish, for example, the genes associated with Bardet-Biedl Syndrome (BBS), whose phenotypes include retinal degeneration, obesity, polydactyly, hypogonadism, and renal dysfunction. Zebrafish with knockdown of BBS genes by morpholinos exhibited gastrulation defects, including a shortened body axis, longer somites, and a kinked notochord.17–20 Based the phenotypes caused by knockdown of individual BBS genes in zebrafish, Zaghloul and colleagues have evaluated 125 putative disease-causing alleles of 14 BBS associated genes. 21 The authors showed that some alleles can fully rescue the morphants' phenotype, some alleles only partially rescue the phenotype, and some alleles yield no rescue, highlighting the genetic complexity of BBS gene mutations. However, a significant fraction of mutations produced phenotypes worse than those of morpholino-knockdown embryos, showing dominant-negative effects. 21 Thus, the zebrafish provides an effective platform to test the functionality of disease alleles.
Zebrafish have also been used to investigate the function of RP-causing genes. Here we provide an overview of the roles of RPGR and RP2, and review zebrafish models for XLRP and functional characterisation of RPGR and RP2 in zebrafish.
Genes of XLRP: RPGR and RP2
The RPGR gene, located in chromosome region Xp21.1, accounts for up to 75% of all XLRP cases. There are two major transcripts of RPGR. The first, RPGRex1–19, contains 19 exons and encodes a predicted 90 kDa protein ending in an isoprenylation anchorage signal. The other transcript, RPGRORF15, contains exons 1-14 of RPGRex1-19 plus an unusual carboxy terminal domain exon called ORF15.22–24 The N-terminal region of both transcripts is structurally similar to the protein RCC1, a guanine nucleotide exchange factor (GEF) that activates the small GTPase Ran. 25 Recent data showed RPGR acts as GEF for the small GTPase, Rab8, a protein involving ciliary protein transport. 26 Exon ORF15 encodes a repetitive glycine and glutamic acid-rich domain of unknown function and a conserved basic C-terminal domain. Exon ORF15 harbors a high frequency of mutations including microdeletions, frameshift, and premature stop mutations. 24 In total, about 300 RPGR mutations have been identified that can give rise to a variety of human retinal dystrophies. Of the known mutations in RPGR, 95% cause X-linked forms of retinitis pigmentosa; 27 3% are for cone-rod dystrophies, cone dystrophies or macular dystrophies;29,30 and 2% of mutations cause syndromal forms of XLRP with hearing loss and primary ciliary dyskinesia (Fig. 2A–D). 23

Mutations in RPGR causing XLRP
RPGR is predominantly localized in rod and cone photoreceptor cells in the connecting cilia and basal bodies, but localization has also been reported in the outer segment of some species. 31 RPGR is not restricted to the retina but is also found in the ciliated airway epithelia and the primary cilium of cultured cells.31,32 RPGR interacts with a number of photoreceptor and ciliary proteins. The N-terminal of RPGR was shown to interact with the delta subunit of rod cyclic GMP phosphodiesterase (PDE6D), a highly conserved protein with a major role in rod phototransduction. 33 RPGR also interacts with the RPGR-interacting protein 1 (RPGRIP1), which is also localized in the photoreceptor connecting cilium. 34 Mutations in RPGRIP1 are one of the causes of Leber's congenital amaurosis. The exact function of RPGRIP1 is unknown, but it is necessary for photoreceptor morphogenesis, for disc formation in the photoreceptor outer segments, 35 and for the proper localization of RPGR. Co-immunoprecipitation studies showed that RPGR interacts with several specific proteins involved in the structure and function of intracellular microtubules, supporting a role for RPGR in microtubular organization and transport between the inner and outer segments of photoreceptors. 36
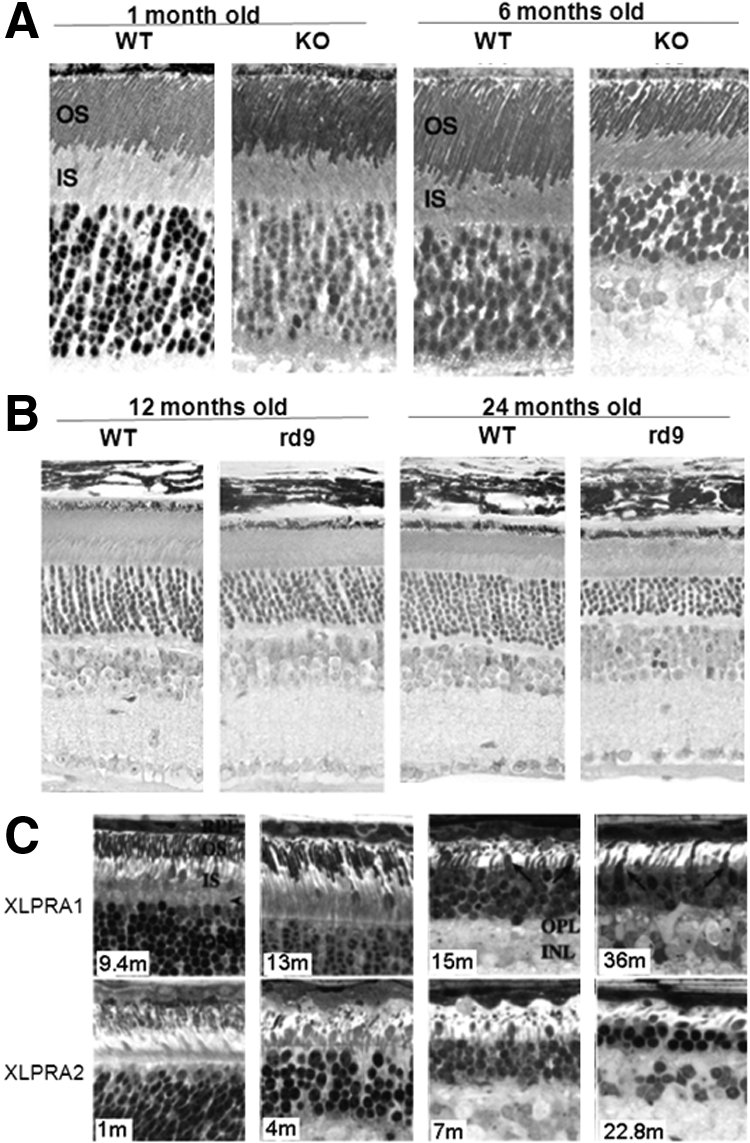
Knockdown of RPGR in the telomerase-transformed human retinal pigment epithelium cell line (hTERT-RPE1) affects ciliogenesis with shortened cilia or a reduced number of cilia, suggesting RPGR has an important role in cilia formation.26,37 Loss of function in human XLRP causes retinal degeneration and mislocalization of rod and cone opsins but normal photoreceptor morphogenesis (Fig. 2E). 38 Knockout (KO) of RPGR in the mouse model produces a slow retinal degeneration with photoreceptor cell loss becoming apparent at 6 months of age (Fig. 3A), but mislocalization of cone opsins was found as early as postnatal day 20. 39 The structure of the axoneme of the connecting cilium appeared well maintained and the KO mouse exhibited well packed discs in the outer segments. However, the newly formed disc membranes at the base of photoreceptor outer segments were notably disorganized in the KO mouse. 21 There are three naturally occurring animal models for XLRP, mouse (rd9) and two canine models. The rd9 mouse carries a 32-base-pair duplication in RPGR exon ORF15, which causes a shift in the reading frame with additional 108 basic residues. Rd9 mice show even slower retinal degeneration than that of RPGR KO mice (Fig. 3B), although both mouse strains show similar disease progression and present mislocalization of cone opsin at an early stage. 40 The two canine models, both with mutations in RPGR exon ORF15, show contrasting genotype-phenotype correlations. Canine XLPRA1 carries a 5-base-pair deletion in exon ORF15, resulting a frameshift and an immediate premature stop codon; while canine XLPRA2 carries a 2-base deletion in exon ORF15, resulting a premature stop codon 71 amino acids downstream with 34 additional basic residues. The XLPRA1 mutant has a slow degeneration of photoreceptors after approximately 6 months of age. In contrast, the XLPRA2 phenotype is very severe with abnormal photoreceptor development and a faster degeneration (Fig. 3C). 41
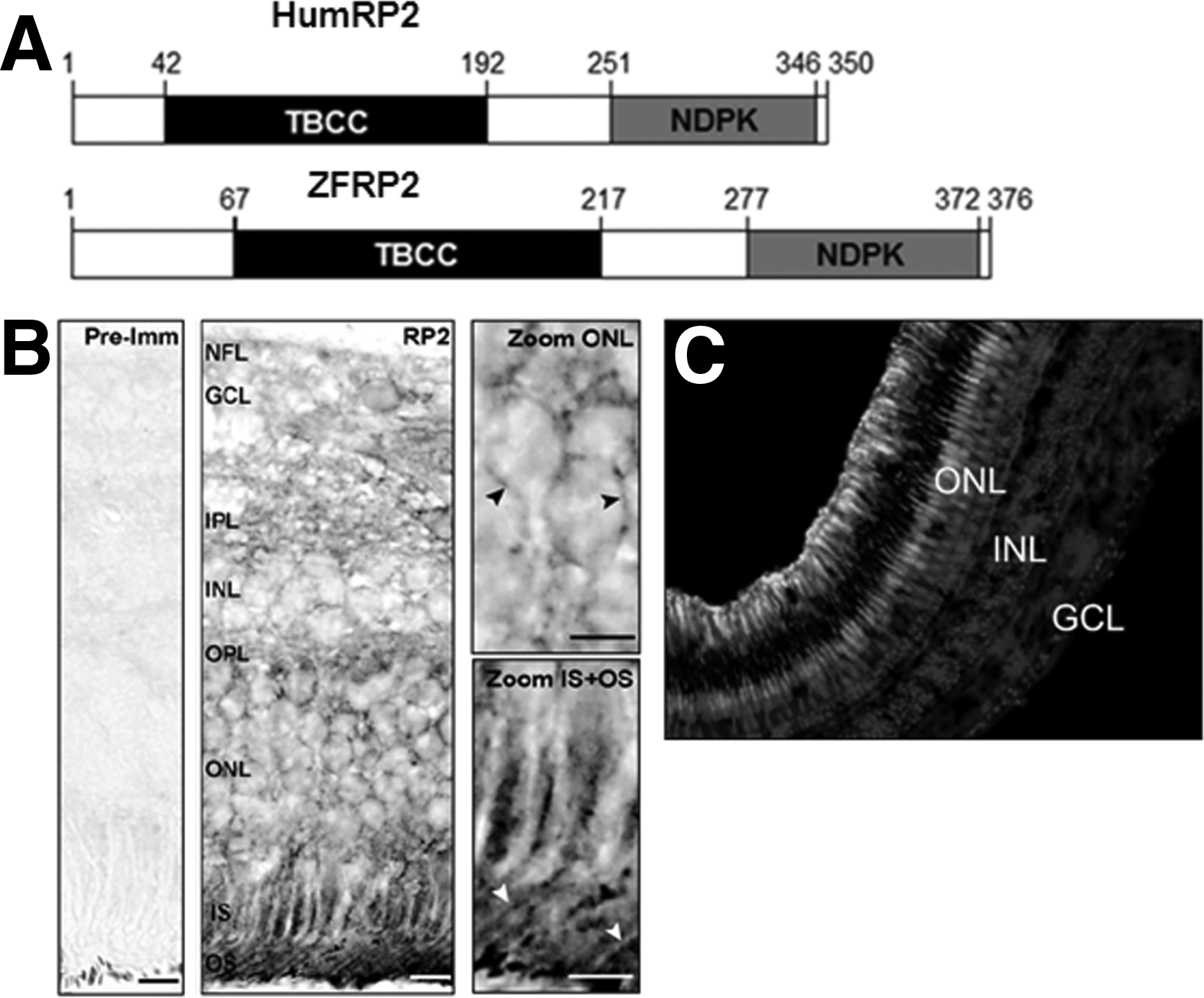
The retinitis pigmentosa 2 (RP2) gene, which was positionally cloned in 1998, accounts for up to 15% of XLRP. 5 RP2 consists of 5 exons, encoding a 350-amino acid protein. The N-terminus of RP2 shares both sequence and structural homology with the tubulin-specific chaperone protein cofactor (TBCC), which is involved in the formation of α/β tubulin heterodimers. The C-terminus consists of a domain with similarity to nucleoside diphosphate kinases (NDPK). 5 All vertebrate RP2 proteins possess an NDPK-like domain, but this domain is not present in RP2 of Ciona and Lancelet. 42 More than 50 mutations have been identified in RP2, most of which are premature stop or frameshift mutations, resulting in truncation proteins. C-terminal truncation mutations, which account for two-thirds of pathogenic RP2 variants, lead to misfolding and subsequent degradation of the resultant nonfunctional proteins. 43 Missense mutations are also found clustered primarily in the TBCC homology domain. Most residues affected by RP-causing missense mutations of RP2 are conserved across species and mutations in the conserved residues tend to cause more severe phenotypes. The pathogenic missense mutation in highly conserved residue Arginine 118 (R118H) does not affect the normal plasma membrane localization but abolishes the tubulin-GAP activity of both RP2 and TBCC. 44
The RP2 protein is widely expressed at low levels in human tissues and does not appear to be enriched in the retina. Previous work has demonstrated RP2 is targeted to the plasma membrane by reversible and irreversible binding of its N-terminal amino acids. 45 The 15 amino acids of the N-terminal appear to be sufficient for targeting RP2 to the membrane. RP-causing mutations at the N terminus were shown to result in failure of the association of RP2 protein with the plasma membrane, indicating that post-translational modifications are critical for the correct localization and function of RP2. 45 RP2 interacts with the ADP ribosylation factor-like protein, Arl3, through its N-terminal β-helix domain and functions as a GTPase activating protein for Arl3. Most of the disease associated RP2 mutations tested exhibited reduced activity of GTP hydrolysis. 46 Recently RP2 has been reported to localize to the structures that regulate the export of proteins from the photoreceptor inner segment to its outer segment, specifically the basal body, the Golgi complex and the pericillary ridge. 47 The targeting of RP2 to the ciliary base depends on N-terminal binding and is regulated by importin β2. 48 Loss of RP2 in cell lines results in the dispersal of the vesicles cycling cargo from the Golgi complex to the cilium, suggesting a specific role for RP2 in Golgi to cilia membrane traffic.46,47 Indeed, it was shown recently that RP2 cooperates with Arl3 to target G protein and NPHP3 to the cilia.49,50
Zebrafish Model for the RPGR Gene
Zebrafish have two RPGR genes, ZFRPGR1 and ZFRPGR2, on chromosomes 9 and 11 respectively, probably due to genome duplication that occurred in teleosts. 51 The ZFRPGR1 gene is homologous to human RPGRORF15 but lacks a transcript similar to human RPGRex1-19, whereas ZFRPGR2 has at least two transcripts, ZFRPGR2ORF15 and ZFRPGR2ex1-17, that are similar to human RPGRORF15 and RPGRex1-19, respectively. ZFRPGR1ORF15 consists of at least 13 exons and encodes a 1697 amino acid protein. ZFRPGR2ORF15 contains 14 exons and is predicted to encode 1413 amino acids. ZFRPGR2ex1-17 contains 17 exons and encodes 708 amino acids. 51 There is a high degree of homology in the RCC1-like domains between human and zebrafish RPGR. Both ZFRPGR1ORF15 and ZFRPGR2ORF15 contain a glutamic and glycine-rich domain similar to that in human RPGRORF15. Between human RPGRex1-19 and ZFRPGR2ex1-17 the isoprenylation signal motif is conserved (Fig. 4A and B). Although ZFRPGR1 and ZFRPGR2 have different developmental and tissue expression patterns, both ZFRPGR1 and ZFRPGR2 have high transcript expression in the eye. Expression of the ZFRPGR2 gene is most similar to the pattern of RPGR in mammals and in Xenopus laevis. 52 In the adult zebrafish eye, both ZFRPGR1 and ZFRPGR2 are expressed in the connecting cilia of rods and cones and in the outer segment of rods, similar to the localization of human RPGR (Fig. 4C–E). 53

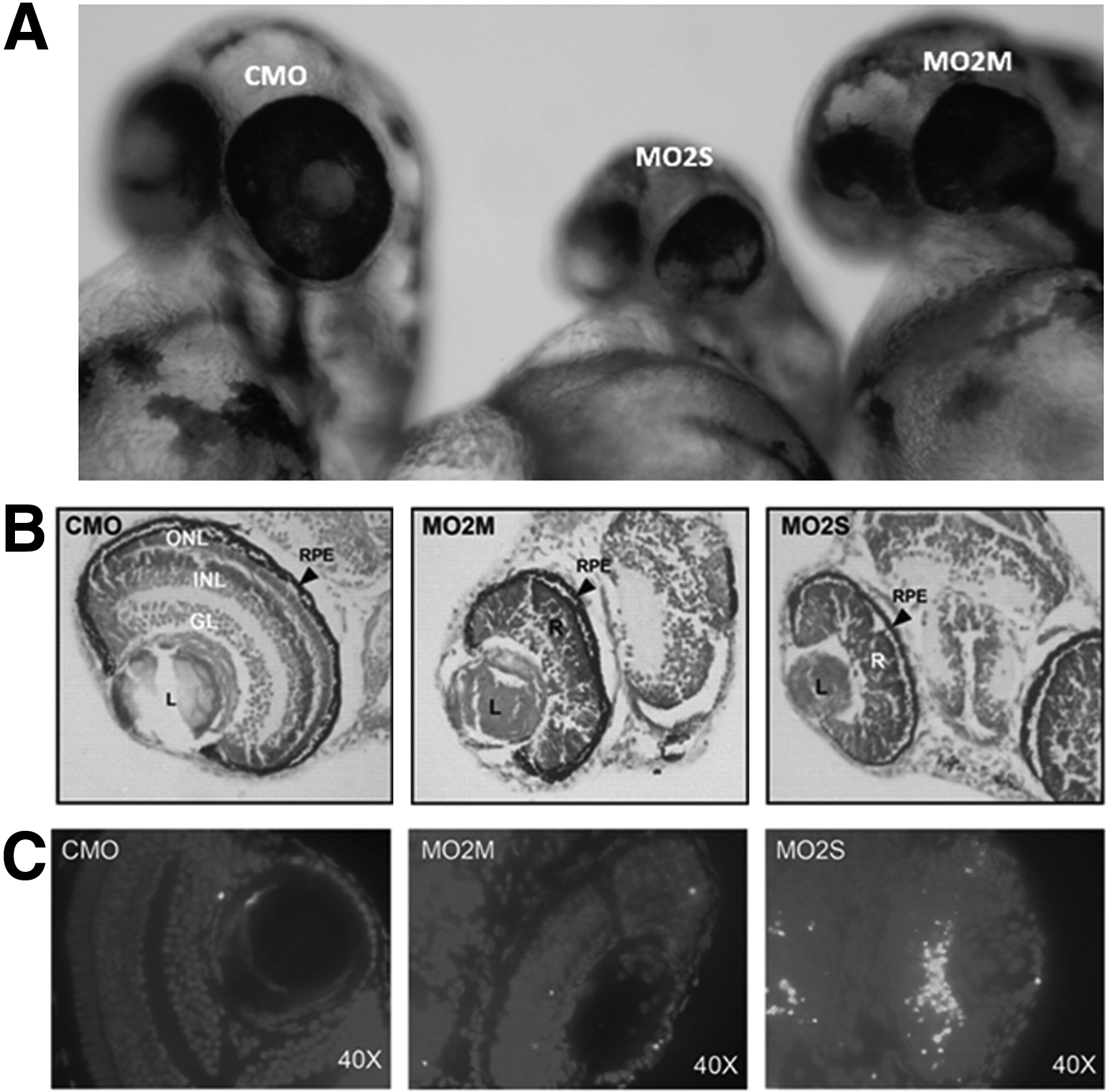
With morpholino knockdown of ZFRPGR1 or ZFRPGR2 in zebrafish, only ZFRPGR2 morphants have developmental defects that range from small eyes to convergent extension defects, consistent with a ciliary phenotype. Depletion of ZFRPGR2 led to both defective lamination of the retinal cell layers and abnormal photoreceptors with absent outer segments at 72 hpf as a result of an ongoing increase in apoptotic cell death in the retina (Fig. 5). Those defects could be rescued by human RPGRORF15 but not by RPGR ex1-19 or by RP-causing mutations. 51 Ghosh et al. 54 knocked-down zebrafish RPGR (ZFRPGR2) with injection of translation-blocking morpholinos and found that the ZFRPGR2 morphants had significantly reduced length of Kupffer's vesicle cilia, associated with ciliary dysfunction. The morphants did not show any abnormality in retinal development. The phenotype of ciliary dysfunction could be rescued by injecting human RPGR mRNAs. Three RP-associated missense mutations (G60V, G165V, and G173R) in the RCC1-like domain (RCCL) can also rescue the ZFRPGR2-knockdown phenotypes, suggesting hypomorphic effects of the mutations. Four nonsense mutations (T124X, K190X, L280X and E589X), a missense mutation (T99N), and three truncation mutations (T124fs, K190fs and L280fs) in RCCL did not rescue the ciliary defect phenotype, suggesting loss-function effects. Two nonsense mutations in exon ORF15 could rescue the phenotypes, suggesting the truncated proteins have some function. However our study did not show that truncated mutations in exonORF15 could rescue ZFRPGR2 knockdown phenotypes. 51 Gerner and associates 55 have also identified a zebrafish RPGR ortholog (the same as ZFRPGR1ORF15) and showed that its depletion caused developmental changes, including an abnormal and shortened body curvature, smaller eyes, a thin mid-to-hind brain boundary, and hydrocephalus. About 6% of morphants also have renal cysts. However, in our study, ZFRPGR1ORF15 suppression did not produce any abnormality in phenotype throughout early development. 51 The different phenotypes of RPGR morphants shown in ours and other studies are possibly caused by different efficiency of RPGR knockdown.
RPGR has been suggested to have a role in vesicle transport by association with both retrograde (cytoplasmic dynein–dynactin complex) and anterograde (kinensin-2) molecular motors. 43 Zebrafish has been used to monitor pigment granule (melanosome) movement along microtubules within nonciliated pigmented skin cells (melanophores). Bi-directional translocation of melanosomes towards the cell center (aggregation) or towards the cell periphery (dispersion) is carried out by different motor proteins. The aggregation of pigment granules requires a minus-end-directed molecular motor protein (cytoplasmic dynein), whereas dispersion requires a plus-end-directed motor protein (kinesin-2). ZFRPGR2 morphants have a significant delay in the aggregation, but not dispersion, of melanosomes. 51 These results suggest that RPGR has a role in the retrograde transport of zebrafish melanosomes mediated by cytoplasmic dynein.
Zebrafish Model for RP2
Zebrafish only have one copy of RP2 on chromosome 6. The zebrafish RP2 transcript (ZFRP2) encodes a 376 amino acid protein and contains at least five exons, encompassing approximately 10.2 kb of DNA. The deduced amino acid sequence of ZFRP2 is highly homologous to those of other fish species (81% identity to the Fugu fish and 78% identity to stickleback) and has 65% identity to human RP2. 56 Similar to human RP2, ZFRP2 also has a N-terminal region of homology to TBCC and a C-terminus with similarity to NDPK (Fig. 6A). The proposed motif for reversible and irreversible binding at the N-terminal of RP2, which is necessary for both trafficking of RP2 to the plasma membrane and to ciliary targeting, is conserved between humans and zebrafish.56,57 ZFRP2 is expressed in oocytes, early cleavage stage embryos, and at later stages of development. Expression of ZFRP2 was detected in all analyzed adult tissues and not enriched in the retina, consistent with the expression patterns of human and mouse RP2. 5 Within the retina, the localization of ZFRP2 protein is also similar to that of the human and the mouse, as it is present in both rod and cone photoreceptors extending from the tip of the outer segment to the inner segment and the synaptic terminals (Fig. 6B and C).56,58
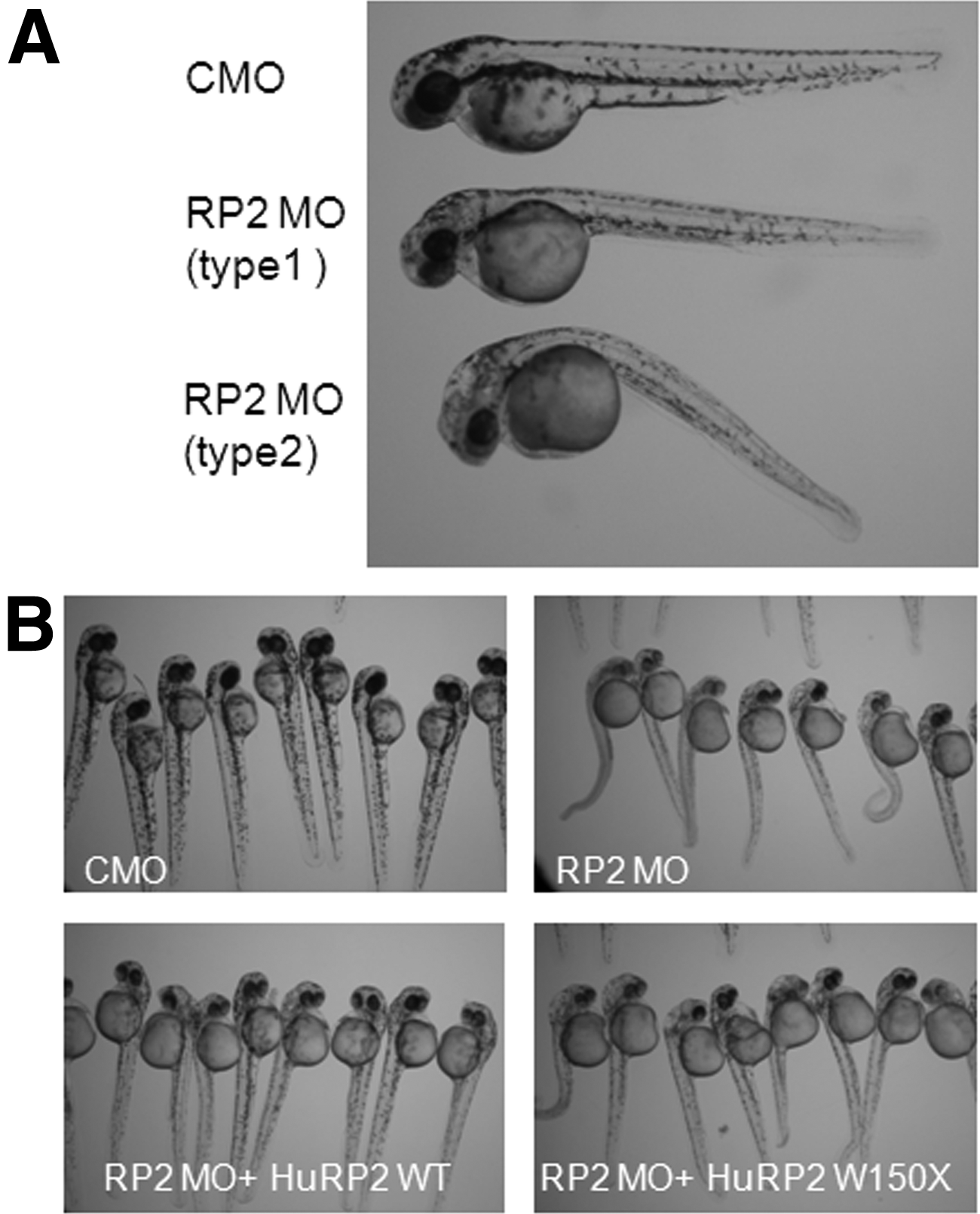
Morpholino-mediated knockdown of ZFRP2 caused a mild developmental delay, ciliary dysfunction, and multiple developmental defects, including body curvature, hydrocephalus, and pericardial effusion. 57 Approximately 20% of RP2 morphants showed a cystic kidney phenotype. The defects of left-right symmetry and heart looping in RP2 morphants are similar to those exhibited in polycystin 2 morphants.59,60 Co-knockdown of both RP2 and polycystin 2 increases the heart looping defect, suggesting that RP2 and polycystin 2 share a common functional pathway. 57 Depletion of ZFRP2 also caused a retinal defect, ZFRP2 morpholino-injected embryos exhibited abnormal dark brown and nontransparent eyes as early as 24 hpf, and had small eyes and small heads with curved bodies and tails at 48 hpf. ZFRP2-deficient morphants displayed defective retinal lamination without discrete ganglion cell, inner nuclear, or outer nuclear layers. The small eye phenotype appears with both abnormal retinal development and extensive retinal cell death, and can be partially rescued by human wild-type RP2 and by two RP-associated missense mutations (R118H and E138G), but not by any of five other missense mutations (C86Y, P95L, C108G, R120K, and L253R) or by a nonsense mutation (W150X) which cause RP (Fig. 7).56,61
Conclusion
The zebrafish have become an increasing attractive model for undersatnding the pathogenesis of human diseases. This model provides us with a unique opportunity to directly address the function of gene mutations in the human XLRP disease. Recent studies demonstrate a surprising degree of functional conservation between human XLRP-causing genes and their zebrafish orthologues. Morpholino-mediated knockdown of the XLRP-causing RPGR gene or the RP2 gene results in defects of the cillia of the retina and throughout the body, suggesting that both RPGR and RP2 are involved in ciliary structure and functions. Human RPGR and RP2 mRNA can rescue the phenotypes of zebrafish morphants, providing a solid platform to test RP-causing mutants before proposing XLRP gene therapy trials.
Footnotes
Acknowledgment
This work was supported by the Royal Society London, the TENOVUS Scotland, the WH Ross Foundation, the National Eye Research Centre, the Rosetrees Trust, the Visual Research Trust, the UK Fight for Sight and the Carnegie Trust for the Universities of Scotland.
Disclosure Statement
No competing financial interest exist.