
Abstract
Abstract
Synthetic targeted endonucleases such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) have recently emerged as powerful tools for targeted mutagenesis, especially in organisms that are not amenable to embryonic stem cell manipulation. Both ZFNs and TALENs consist of DNA-binding arrays that are fused to the nonspecific FokI nuclease domain. In an effort to improve targeted endonuclease mutagenesis efficiency, we enhanced their catalytic activity using the Sharkey FokI nuclease domain variant. All constructs tested display increased DNA cleavage activity in vitro. We demonstrate that one out of four ZFN arrays containing the Sharkey FokI variant exhibits a dramatic increase in mutagenesis frequency in vivo in zebrafish. The other three ZFNs exhibit no significant alteration of activity in vivo. Conversely, we demonstrate that TALENs containing the Sharkey FokI variant exhibit absent or severely reduced in vivo mutagenic activity in zebrafish. Notably, Sharkey ZFNs and TALENs do not generate increased toxicity-related defects or mortality. Our results present Sharkey ZFNs as an effective alternative to conventional ZFNs, but advise against the use of Sharkey TALENs.
Introduction
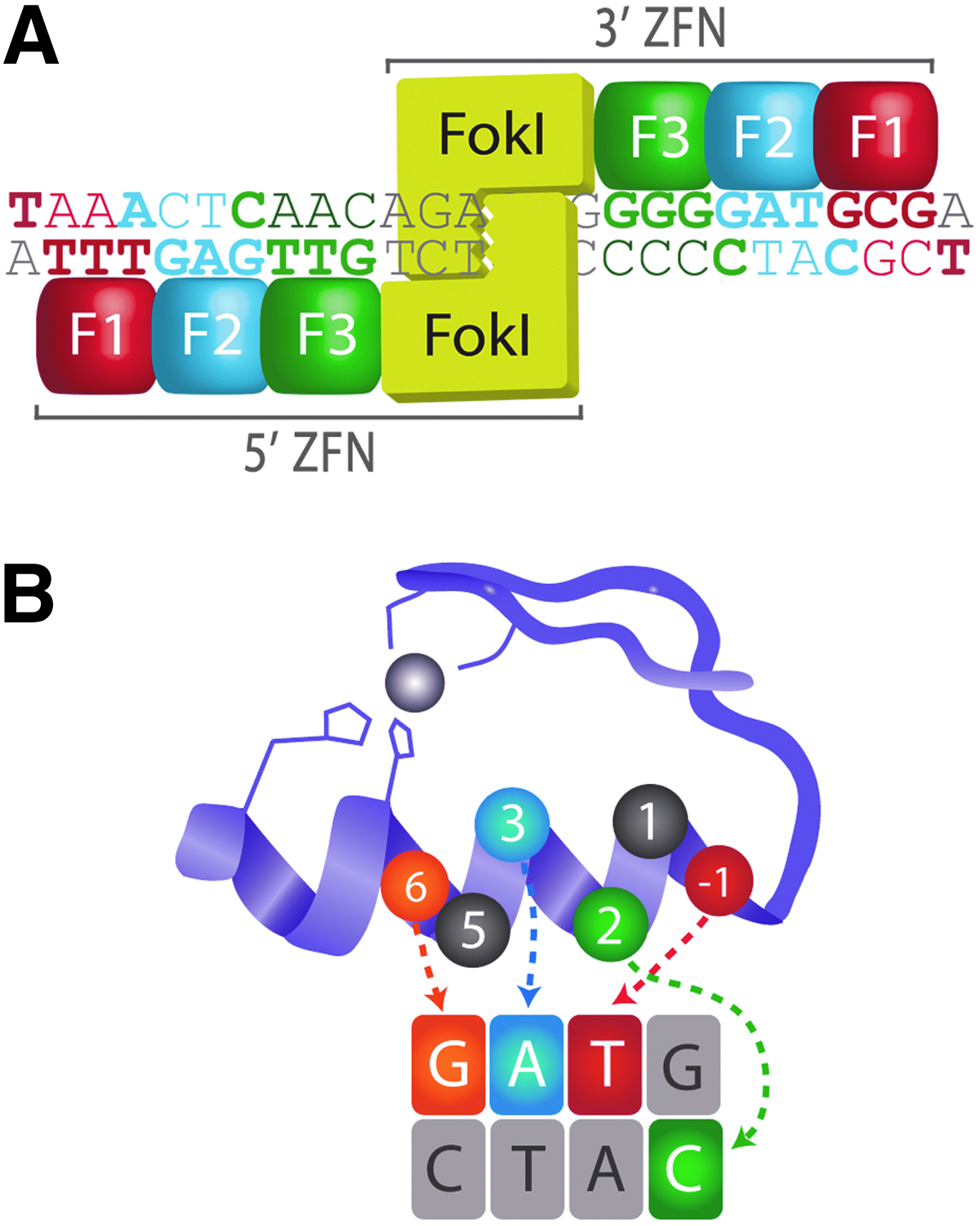
Zinc finger nuclease (ZFN) structure and target-site recognition.
Individual zinc fingers use a seven-amino acid motif to recognize and bind a specific DNA triplet sequence, with possible additional contact to the fourth base on the opposite strand (Fig. 1B).49,50 This motif can be modified to generate custom zinc finger domains with novel DNA sequence specificities. 51 Precise recognition of ZFN target sequences is achieved by arranging zinc fingers with desirable DNA-binding specificities in tandem arrays. The mutagenesis activity of a ZFN is primarily dependent on the DNA-binding affinity and specificity of its zinc finger array.11,52 Consequently, multiple approaches have been used to generate custom zinc finger arrays. The modular assembly approach makes use of individual, pre-selected zinc finger domains that are assembled into arrays.51,53–58 Although rapid and facile, this strategy does not take context-dependent effects between neighboring zinc fingers into consideration,59,60 and it exhibits a high rate of failure in mutagenesis applications.28,61,62 Cell-based library screening methods have been successfully used to construct and identify zinc finger arrays with desirable sequence specificities.21,45,47,52,63–66 However, library construction and validation is time- and labor intensive. Furthermore, the success rate for obtaining mutations using arrays identified through cell-based screening methods is still somewhat low (∼50%–67%).21,30,32,46,67
Transcription activator-like effector nucleases (TALENs) are a second technology that is used for targeted mutagenesis applications. To date, TALENs have been successfully used to mutagenize mammalian cell lines,68–75 plants,69,76–78 yeast, 79 Drosophila melanogaster,72,80 nematodes, 81 silkworm,82,83 Xenopus laevis,72,84,85 mice,86–88 rats,88–90 swine 91 medaka, 92 and zebrafish.72,93–99 Similar to ZFNs, TALENs also induce double-strand breaks that are repaired through homologous recombination, or NHEJ to generate insertions and deletions that alter gene function. A TALEN consists of a transcription activator-like (TAL) effector array that is fused to the FokI endonuclease domain.100,101 TAL effectors recognize and bind to specific DNA sequences through series of repeated modules.102–105 Each module contains a repeat variable di-residue that preferentially recognizes and binds to a specific nucleotide (C, T, A, or G/A).103–105 Consequently, TALENs can be engineered to recognize nearly any DNA sequence, without the requirement for selection assays. Notably, TALENs have been shown to elicit a greater mutation rate than ZFNs in zebrafish. 106 However, the somatic mutation rate obtained by using TALENs is still quite variable (<1% to 100%), and it depends on the selected TALEN scaffold, as well as the targeted locus. 95
We sought to improve the efficiency of ZFN and TALEN synthetic targeted endonucleases for use in zebrafish mutagenesis. Toward this aim, we examined the activity of both ZFNs and TALENs containing a FokI nuclease variant termed Sharkey. 107 We demonstrate that all tested Sharkey ZFNs exhibit greater in vitro cleavage of target-site DNA than controls. However, only one out of four Sharkey ZFNs displays significantly greater activity in vivo in zebrafish, producing a higher frequency of insertion/deletion mutations than control ZFNs. As with ZFNs, we demonstrate that Sharkey TALENs exhibit greater in vitro cleavage of target-site DNA than controls. However, all Sharkey TALENs examined fail to produce any insertion/deletion mutations in zebrafish, displaying absent or significantly reduced in vivo mutagenic activity in comparison to control TALENs. Notably, embryos injected with Sharkey ZFNs and TALENs do not exhibit an increase in toxicity-related defects or mortality.
Materials and Methods
Ethics statement
Embryonic and adult AB strain zebrafish were cared for in accordance with the Canadian Council for Animal Care guidelines. This study was approved by the University of Alberta Animal Care and Use Committee for Biosciences (protocol 427).
Design and construction of ZFN arrays
Algorithm-based (http://pgfe.umassmed.edu/ZFPsearch.html) detection of suitable ZFN target sequences was performed as previously described,
47
with preference given to purine-rich sequences, guanine-rich sequences, and sequences containing a six-nucleotide spacer region between ZFN recognition sites. The following ZFN target sequences were chosen for bacterial one-hybrid selection (ZFN recognition sites underlined):
prp2, 5′-C hmx4, 5′-T crx, 5′-A
To identify and construct suitable ZFN arrays for prp2, hmx4, and crx, zinc finger library construction and two-stage omega-based bacterial one-hybrid selection of zinc finger arrays was performed as previously described, 47 except for hmx4 ZFNs, in which the bacterial one-hybrid selection target-site plasmids were modified so that the fourth base pair on the opposite strand always corresponded to the hmx4 zinc finger domain's recognition site (as opposed to the Zif268 scaffold zinc finger domain's recognition site) (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/zeb). This was done to increase the context-dependent selection of individual zinc finger domains that bind to the hmx4 target-site. Furthermore, the degeneracy of hmx4 library primers was manually altered to encode an increased number of zinc finger protein motifs, which were previously shown to recognize triplets present within the hmx4 target-site (Supplementary Table S2). 21 Refer Supplementary methods for a comprehensive protocol detailing bacterial one-hybrid ZFN array selection. The seven-amino-acid target-site recognition motifs of each ZFN chosen for analyses in vivo are listed in Supplementary Table S3.
The prp1, nlz2, gdf11, and sfrp5 ZFN arrays were designed through context-dependent assembly (CoDA) as previously described,
28
using the ZiFiT Targeter Version 3.3 algorithm (zifit.partners.org/ZiFiT) to select the ZFN target-sites.108,109 Preference was given to purine-rich sequences, guanine-rich sequences, and sequences containing a six-nucleotide spacer region between ZFN recognition sites. The following ZFN target sequences were chosen:
prp1, 5′-G nlz2, 5′-G gdf11, 5′-A sfrp5, 5′-T
For prp1, information from the algorithm was then used to manufacture a custom double-stranded minigene in pIDTSMART-KAN (IDT). For nlz2, gdf11, and sfrp5, ZFN arrays were constructed through overlapping PCR as previously described. 47 Refer Supplementary Table S4 for the seven-amino-acid target-site recognition motifs of each CoDA zinc finger array.
ZFN constructs
All selected zinc finger arrays were cloned into the previously described pCS2-HA-GAAZFP-FokI-RR (5′ arrays) or pCS2-Flag-TTGZFP-FokI-DD (3′ arrays) ZFN expression vectors (Addgene). 47 The DD (R487D, N496D) and RR (D483R, H537R) cleavage domain mutations favor heterodimeric cleavage activity, and, therefore, reduce ZFN toxicity.22,110
ZFNs containing the Sharkey FokI cleavage domain have previously been shown to exhibit greater in vitro activity than those containing the wild-type FokI cleavage domain.
107
Sharkey variants of the heterodimeric ZFN expression vectors mentioned earlier (pCS2-Flag-TTGZFP-SharkeyFokI-DD; pCS2-HA-GAAZFP-SharkeyFokI-RR) were synthesized through site-directed mutagenesis of the original expression vectors using the following primers (altered nucleotide sequence underlined):
(S418P-F), 5′-ATTGAATTAATTGAAATTGCCAGAAAT (S418P-R), 5′-CAAGAATTCTATCCTGAGTGG (K441E-F), 5′-GAAAGTTTATGGATATAGAGGT (K441E-R), 5′-TTCCTTGATCCACCCAAATGTT
All constructs were confirmed by Sanger sequencing.
Design and construction of TALENs
All TAL effector arrays were designed essentially as previously described,
69
using the TAL Effector Nucleotide Targeter 2.0 (https://tale-nt.cac.cornell.edu) to select the TALEN target sites.
111
Preference was given to sequences occurring on or near the translation start site of targeted genes. The following TALEN target sequences were chosen:
wwtr1, 5′- hoxb1b, 5′-
Assembly of custom TAL effector arrays was performed essentially as previously described. 69 All TAL Effector arrays were cloned into the previously described pCS2TAL3-RR or pCS2TAL3-DD TALEN expression vectors (Addgene). 96 Sharkey variants of the heterodimeric ZFN expression vectors mentioned earlier (pCS2TAL3-SharkeyFokI-RR; pCS2TAL3-SharkeyFokI-DD) were synthesized through site-directed mutagenesis of the original expression vectors using the same mutagenic primers used to make the heterodimeric Sharkey ZFN expression constructs. Constructs were confirmed by Sanger sequencing.
Targeted endonuclease protein synthesis and in vitro DNA cleavage assay
Our in vitro DNA cleavage assay was adapted from the in vitro transcription-translation assay for rapid screening of ZFNs for sequence-specific cleavage activity. 5 ZFN or TALEN constructs were transcribed and translated using the TNT SP6 Coupled Rabbit Reticulocyte Lysate System (Promega) from 100 to 500 ng of pCS2-FokI ZFN (RR or DD; control or Sharkey) or pCS2TAL3 TALEN (RR or DD; control or Sharkey) plasmid DNA according to the manufacturer's specifications. After synthesis, 0.1–2 μL of 5′ ZFN or TALEN (RR; control or Sharkey) and 3′ ZFN or TALEN (DD; control or Sharkey) protein lysates were combined with 500 ng of target-site-containing plasmid DNA (pCR4-TOPO for hmx4 and wwtr1 constructs; pBSK for prp1; pCR2.1-TOPO for prp2), 2 μL 10×Restriction Buffer 4 (NEB) or 10×FastDigest Buffer (Fermentas), and 0.5 μL of plasmid-linearizing restriction enzyme (NcoI for prp2, hmx4, crx and wwtr1; SalI for prp1) (20 μL total volume). This reaction was incubated at 37°C for 2 (prp2, hmx4, crx, and wwtr1) to 3.5 (prp1) hours. Digested plasmid DNA was purified using a gel-extraction kit (QIAquick Gel Extraction Kit, QIAGEN; GeneJET Gel Extraction Kit, Fermentas), and digested DNA fragments were analyzed by agarose gel electrophoresis. Anecdotal evidence suggests that the plasmid purification step improves the resolution and separation of DNA fragments.
Western analysis
In vitro translated crude protein lysates (2 μL/sample) were run out on NuPAGE Novex Bis-Tris gels using the XCell Sure Lock Mini-Cell system (Invitrogen) according to the manufacturer's specifications. Proteins were transferred to polyvinylidene difluoride membrane using a TE 77 enhanced chemiluminescence (ECL) Semi-dry transfer apparatus (Amersham). Membranes were blocked in 5% skim milk in Tris-buffered saline with 0.1% Tween-20 (TBST) (anti-Flag antibody) or 5% skim milk with 2% bovine serum albumin (BSA) and 2% goat serum in TBST (anti-HA antibody). Membranes were incubated in either anti-Flag antibody (Sigma; 1:2,000 dilution in TBST with 5% skim milk) or anti-HA antibody (GenScript, Sigma or Covance; 1:1,000 or 1:2,000 dilution in TBST with 2% BSA and 2% goat serum). After TBST washes, both membranes were incubated in 1:5,000 or 1:7,500 secondary antibody (goat anti-mouse HRP FAB fragments from Amersham) in TBST. After TBST washes, an ECL reaction was performed using PicoSignal (Pierce).
mRNA synthesis
All mRNAs were transcribed from NotI-linearized, purified pCS2-FokI or SalI-linearized, purified pCS2TAL3 (RR or DD; control or Sharkey) plasmid DNA templates using the SP6 mMessage mMachine kit (Ambion) according to the manufacturer's specifications, and were purified using Amicon Ultra 50K centrifugal filters (Millipore) or lithium chloride precipitation. mRNA concentration was determined via spectrophotometry, and mRNAs were diluted in diethyl-pyrocarbonate-treated water.
Zebrafish husbandry and mutagenesis
Embryonic and adult AB strain zebrafish were cared for according to standard protocols. 112 Embryos were grown at 28.5°C in embryo media and staged according to standardized morphological milestones. 113
The following amounts of 5′ and 3′ control or Sharkey ZFN mRNAs were injected into single-cell zebrafish embryos: prp2 ZFN mRNA, 20 pg; hmx4 ZFN mRNA, 200 pg; crx ZFN mRNA, 100 pg; and prp1 ZFN mRNA, 100 pg. The dosage of each injected ZFN mRNA was titrated and chosen to minimize embryonic toxicity and lethality (producing 65%–75% ‘normal’ embryos for control ZFN mRNAs). For each category of injected embryos, genomic DNA was isolated from pooled 24 h post-fertilization embryos, essentially as previously described. 114 PCR fragments containing the ZFN recognition site were amplified and cloned into pCR4-TOPO (hmx4, crx), pCR2.1-TOPO (prp2) or QIAGEN pDrive Cloning Vector (prp1). The cloned product was used to transform TOP10 cells (Invitrogen) or EZ Competent Cells (QIAGEN). After transformation, 96 independent colonies from each sample were picked, and from these the cloned PCR product was amplified using Taq DNA polymerase. The amplified clones were sequenced using M13R (prp2, hmx4, and crx) or T7 (prp1) primer to determine the frequency and type of mutations present at the ZFN target site. Significant differences in mutation frequencies among embryos injected with Sharkey versus control ZFNs were determined using two-tailed Fisher's exact tests.
The following amounts of 5′ and 3′ control or Sharkey TALEN mRNAs were injected into single-cell zebrafish embryos: wwtr1 TALEN mRNA, 200 pg; hoxb1b TALEN mRNA, and 400 pg. For each category of injected embryos, genomic DNA was isolated from pooled 24 h post-fertilization embryos, essentially as previously described. 114 PCR fragments containing the ZFN recognition site were amplified and cloned into pCR4-TOPO. The cloned product was used to transform TOP10 cells (Invitrogen). After transformation, 90 independent colonies from each sample were picked and diluted in 25 μL of water. High-resolution melt curve analysis (HRMA) was performed on each sample to detect TALEN-induced mutations. Each 10 μL of HRMA reaction contained the following: 2 μL of diluted plasmid DNA, 5 μL of MeltDoctor HRM Master Mix (Applied Biosystems), 0.6 μL each of 5 μM forward and reverse primer, and 1.8 μL of water. Each reaction was run using the 7500 Fast Real-Time PCR System (Applied Biosystems), with the following PCR cycle conditions: initial denaturation at 95°C, 10 min; 40 cycles of amplification {95°C, 15 s 60°C, 20 s}; disassociation curve at 95°C, 15 s; 60°C, 1 min; 95°C, 15 s; and 60°C, 15 s. Melting profiles were analyzed using HRM v.2.0 software (Life Technologies). Representatives of detected variants were sequenced using M13R or M13F primer to determine the frequency and type of mutations present at the TALEN target site. Significant differences in mutation frequencies among embryos injected with Sharkey versus control TALENs were determined using two-tailed Fisher's exact tests.
Analyses of targeted endonuclease toxicity
The following amounts of 5′ and 3′ control or Sharkey ZFN mRNAs were injected into single-cell zebrafish embryos: prp2 ZFN mRNA, 20 pg; hmx4 ZFN mRNA, 200 pg; crx ZFN mRNA, 100 pg; and prp1 ZFN mRNA, 100 pg. 200 pg each of 5′ and 3′ control or Sharkey wwtr1 TALEN mRNAs were injected into single-cell zebrafish embryos. At ∼24 h post-fertilization, embryos were categorized according to morphological phenotype (normal, ‘monster’, which indicates any morphological phenotype that differs from wild type, or dead) and counted. The morphological phenotypes of control, uninjected embryos were likewise assessed. Each set of injections/assessment was conducted in triplicate. Significant differences in morphology and lethality among treatments were determined using two-tailed Fisher's exact tests with Bonferroni correction on pooled datasets.
Results
Rapid in vitro verification of ZFN target-sequence cleavage
We used two distinct methods to engineer ZFNs that selectively bind target sequences within seven chosen zebrafish genes: prion protein 2 (prp2/prnprs3), H6 family homeobox 4 (hmx4), cone-rod homeobox (crx), prion protein 1 (prp1/prnprs1), secreted frizzled-related protein 5 (sfrp5), growth differentiation factor 11 (gdf11), and nocA-like zinc finger protein 2 (nlz2/znf503). For prp2, hmx4, and crx, we used a previously described bacterial one-hybrid system to screen a diverse library of zinc finger domains for their ability to bind the selected target-sequence. 47 For prp1, sfrp5, gdf11, and nlz2, we used a CoDA method that makes use of pre-selected zinc finger arrays. 28 All zinc finger arrays were cloned into pCS2-FokI ZFN (DD and RR) expression vectors. 47 The DD and RR cleavage domain mutations prevent homodimeric cleavage activity, thereby reducing ZFN off-target activity and toxicity.22,66,110
Previous studies used restriction, PCR, or sequencing-based genotyping assays to analyze somatic mutations in ZFN-injected zebrafish embryos.28,46,47 Such studies demonstrate that mutagenesis success rates are variable, and are highly dependent on target-site sequence and ZFN array construction methodology. Our bacterial one-hybrid screens for prp2, hmx4, and crx yielded a large variety of prospective 5′ and 3′ ZFN arrays (data not shown). However, the laborious nature of identifying in vivo somatic mutations precludes the analysis of multiple potential ZFN arrays. Consequently, before testing individual pairs of ZFNs in vivo, we assayed their ability to cleave their respective gene's target sequence in vitro. To do this, we used a modified in vitro transcription-translation assay. We synthesized 5′ and 3′ ZFN crude protein lysates by coupled in vitro transcription/translation, and incubated them along with plasmid containing the ZFN target-sequence, buffer, and plasmid-linearizing restriction enzyme. An analysis of purified, digested plasmid DNA by gel electrophoresis demonstrates whether or not ZFN arrays possess appropriate DNA cleavage activity. For prp2, hmx4, crx, and prp1, we selected those ZFNs with the greatest in vitro cleavage activity for subsequent analyses (data not shown). Notably, the sfrp5, gdf11, and nlz2 CoDA 5′ and 3′ ZFNs failed to cleave their respective targets (data not shown), and were excluded from further analyses.
Increased efficiency of Sharkey FokI nuclease-containing ZFNs in vitro
ZFN efficiency is dependent on the affinity and specificity of individual ZFN arrays,11,52 length and identity of the spacer region between ZFN recognition sites,7,115 interaction between FokI nuclease domains,22,110 and catalytic activity of the FokI nuclease domain. 107 In an attempt to improve the efficiency of our ZFN arrays, we made use of a FokI nuclease variant termed Sharkey. This variant was initially developed and identified through a directed-evolution strategy, and it demonstrates >15-fold more catalytic activity than wild-type FokI nuclease. 107
We first wanted to determine whether incorporating the Sharkey FokI nuclease into our ZFN arrays enhanced their function in vitro. To do this, we used our in vitro DNA cleavage assay to evaluate the abilities of control versus Sharkey FokI nuclease-containing ZFNs to cleave target-site-containing plasmid DNA (Fig. 2). To compare ZFN cleavage activities, we diluted protein lysates (0.5× or 0.1×). As shown for prp2 ZFN pairs (Fig. 2A), samples containing Sharkey prp2 ZFN protein lysate demonstrate more cleavage of prp2 ZFN target-site-containing plasmid DNA than samples containing control prp2 ZFN protein lysate. We observe similar results for hmx4 (Fig. 2B), prp1 (Fig. 2C), and crx ZFNs (Fig. 2D). Western analysis confirmed that comparable amounts of control and Sharkey 5′ and 3′ ZFN proteins were present in the lysates used for these analyses (Supplementary Fig. S1). Our results demonstrate that Sharkey ZFNs exhibit increased in vitro cutting efficiency compared with control ZFNs. Combined, these data suggest that Sharkey FokI nuclease-containing ZFNs cleave DNA more efficiently than control FokI-nuclease containing ZFNs in vitro.
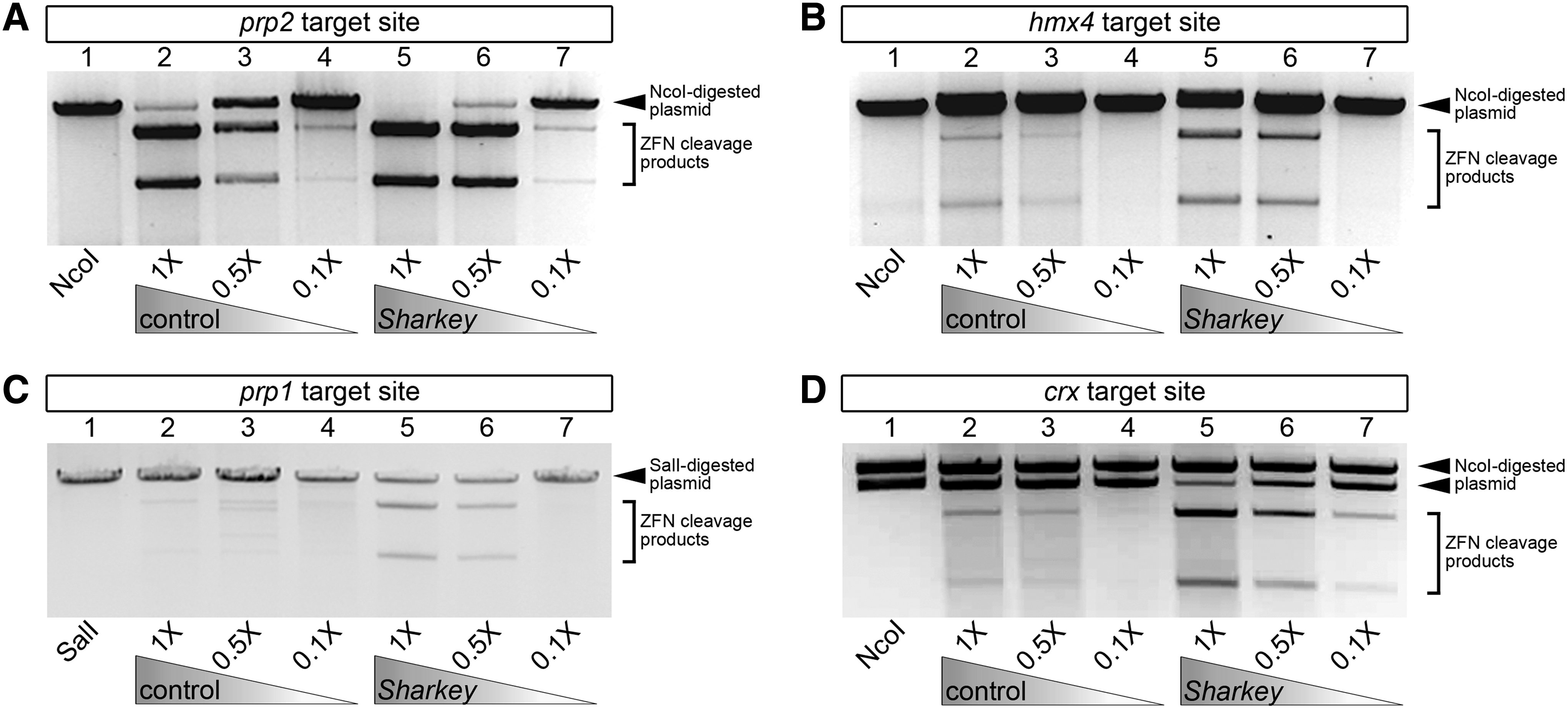
In vitro comparison of target-site specific DNA cleavage activity between control and Sharkey ZFNs. ZFN crude protein lysates were used at normal concentration (1×), or were diluted to one-half (0.5×) or one-tenth (0.1×) the amount. Gel-electrophoretic analyses of prp2
In vivo mutagenesis by Sharkey FokI nuclease-containing ZFNs
Given that Sharkey ZFNs function more efficiently than control ZFNs to cleave target-site DNA in vitro, we next wanted to determine whether Sharkey ZFNs possess more in vivo mutagenic activity than control ZFNs. To do this, we injected single-cell zebrafish embryos with mRNAs encoding control or Sharkey ZFNs, and determined the sequence of target-site genomic DNA. Sequencing results from control versus Sharkey ZFN mRNA-injected embryos are summarized in Table 1. For crx, embryos injected with Sharkey ZFN mRNA exhibit a 26 times greater frequency of target-site-specific insertion and deletion (indel) mutations than embryos injected with control ZFN mRNA (Sharkey, 31.2%, control, 1.2%; Table 1). This difference in crx indel frequency is statistically significant (p<0.0001). For prp2, prp1, and hmx4, the difference in indel frequency between embryos injected with Sharkey ZFN mRNA and embryos injected with control ZFN mRNA is not statistically significant (Table 1; p-values: prp2, 0.738; prp1, 0.621; and hmx4, 0.669). Combined, these data suggest that Sharkey FokI nuclease-containing ZFNs have the capacity to exhibit greater in vivo mutagenic activity than control FokI nuclease-containing ZFNs.
PCR fragments containing the ZFN target site were amplified, cloned, and sequenced. Data indicates combined frequency of insertions and deletions (indel frequency). aIndicates significant difference in indel mutation frequency between embryos injected with Sharkey and control ZFNs, as determined by two-tailed Fisher's exact test (p<0.0001).
ZFN, zinc finger nuclease.
Toxicity of Sharkey FokI nuclease-containing ZFNs
One concern with using Sharkey ZFNs is that increased activity of the FokI nuclease might result in additional off-target effects, thereby increasing the morbidity and decreasing the survival of injected embryos. We, therefore, quantified the proportion of embryos that exhibit nonspecific developmental defects (referred to as ‘monster’-like), and the mortality rates of embryos injected with mRNAs encoding control or Sharkey ZFNs (Fig. 3). In all cases examined, we failed to observe a significant difference (corrected p-value=1.000 for each) in the mortality rates of embryos injected with control versus Sharkey ZFN mRNAs (Fig. 3). Furthermore, we note that embryos injected with control and Sharkey ZFN mRNAs exhibit a comparable proportion of monster-like phenotypes (Fig. 3; corrected p-values: prp2, 0.202; hmx4, 1.000; crx, 1.000). Combined, these data suggest that, in comparison to control ZFNs, Sharkey ZFNs do not significantly alter the morbidity and survival of zebrafish embryos.

Effects of injecting control and Sharkey ZFN mRNAs on embryonic morphology and mortality. Graphs demonstrating the mean proportion of embryos with indicated phenotype at ∼24 h post fertilization, after an injection of mRNAs encoding control FokI or Sharkey FokI prp2
Decreased in vivo mutagenesis by Sharkey FokI nuclease-containing TALENs
Given that some Sharkey ZFNs may exhibit increased in vivo mutagenesis activity in zebrafish when compared with control ZFNs, we next wanted to determine whether applying the Sharkey FokI nuclease variant to our WW domain containing transcription regulator 1 (wwtr1/taz) and homeo box B1b (hoxb1b) TALENs would also increase their activity. To do this, we injected single-cell zebrafish embryos with mRNAs encoding control or Sharkey TALENs, and determined the frequency of indel mutations present in target-site genomic DNA using a combination of HRMA and sequencing. Results from control versus Sharkey TALEN mRNA-injected embryos are summarized in Table 2. Embryos injected with control TALENs demonstrate a modest target-site-specific indel frequency (Table 2; wwtr1, 17.2%; hoxb1b, 5.9%). Conversely, in each case examined, embryos injected with Sharkey TALENs fail to exhibit any target-site-specific indel mutations (Table 2; wwtr1, 0.0%; hoxb1b, 0.0%). The difference in wwtr1 indel formation is statistically significant (p<0.0001). Notably, we fail to observe a significant difference in the mortality rates or monster-like phenotypes of embryos injected with control versus Sharkey TALEN mRNAs (Fig. 4A, corrected p-value=1.000 for each).

Comparison of control and Sharkey wwtr1 transcription activator-like effector nuclease (TALEN)s.
PCR fragments containing the TALEN target site were amplified, cloned, and analyzed through a combination of high-resolution melt curve analysis followed by sequencing. Data indicates the combined frequency of insertions and deletions (indel frequency). aIndicates significant difference in indel mutation frequency between embryos injected with Sharkey and control TALENs, as determined by two-tailed Fisher's exact test (p<0.0001).
TALEN, transcription activator-like effector nuclease.
We next wanted to determine whether incorporating the Sharkey FokI nuclease into our TALENs somehow reduced or destroyed their capacity to cleave target-site DNA. We, therefore, used our in vitro DNA cleavage assay to evaluate the abilities of control versus Sharkey FokI nuclease-containing TALENs to cleave target-site-containing plasmid DNA (Fig. 4B). Notably, protein lysate samples containing Sharkey wwtr1 TALENs demonstrate more cleavage of wwtr1 TALEN target-site-containing plasmid DNA than samples containing control wwtr1 TALENs (Fig. 4B). Western analysis confirmed that comparable amounts of control and Sharkey 5′ and 3′ TALEN proteins were synthesized and present in the lysates used for these analyses (Fig. 4C). Combined, these results suggest that Sharkey FokI nuclease-containing TALENs cleave DNA more efficiently than control FokI-nuclease containing TALENs in vitro, but possess absent or reduced in vivo mutagenic activity in zebrafish.
Discussion
ZFNs have been used to generate mutations in organisms that are not amenable to homologous recombination-based genetic modifications,28,38,39,43,44,46,47 and are being evaluated for usage in gene therapies.24,116,117 Improving these molecular tools is, therefore, relevant to both biological and clinical applications. In this study, we have described techniques that may enhance the efficiency and success of constructing ZFNs for use in mutagenesis applications.
The practical application of ZFNs relies on their ability to generate double-strand breaks in specific DNA sequences. Previous research suggests that both target-site affinity/specificity and cleavage activity are critical determinants of ZFN function.8,10,11,13 Without DNA-binding specificity, engineered ZFNs demonstrate low or off-target cleavage activity.10,22,25,110,118 Off-target ZFN activity results in increased cytotoxicity and nonspecific morphological defects. 52 Context-dependent array selection strategies account for cooperativity among zinc finger domains, and permit recognition of the fourth base pair on the opposite strand of the ZFN target-sequence.49,50,60,119–121 These features allow for the selection of ZFNs with greater target-site affinity and specificity than those obtained through modular assembly approaches.61,62 Multiple context-dependent array construction methods have been used with varying degrees of success to construct ZFN arrays for use in mutagenesis applications.
We successfully used two distinct context-dependent array selection methodologies for constructing ZFNs with desirable target-site specificities. The first method was initially used to generate the Wolfe and Lawson kdrl ZFNs, and it uses a two-phase bacterial one-hybrid assay to construct suitable ZFN arrays. 47 We used this bacterial one-hybrid approach to construct ZFN arrays for prp2, hmx4, and crx. For prp2 and crx, we performed target-site selections and constructed ZFN arrays essentially as previously described. 47 For hmx4, however, we increased context-dependent selection of individual zinc finger domains by modifying the initial screening phase to account for recognition of the fourth base on the opposite strand. We also altered the hmx4 libraries to include OPEN-method zinc finger domain motifs that were previously shown to recognize triplets in the hmx4 target-site. 21 Our prp2, hmx4, and crx ZFNs each function effectively in vivo in zebrafish, generating insertion/deletion (indel) mutations at a detectable frequency in injected embryos (prp2: 4.6%; hmx4: 1.2%; crx: 1.2%).
The second method that we used to construct ZFN arrays is CoDA. The CoDA approach to ZFN array construction combines pre-selected zinc finger pairs into three-finger arrays using standard cloning techniques. 28 CoDA takes context-dependent effects between adjacent zinc fingers into consideration, and it has been successfully used to mutagenize both plants and zebrafish. 28 CoDA target-site identity is quite stringent, owing primarily to the existence of only 18 fixed middle zinc finger units. 28 Nonetheless, we identified an ideal CoDA-compatible target site in the prp1 gene, and used this method to construct prp1 ZFNs. Such an approach is rapid, and with commercial minigene technology, it is not labor intensive. Our prp1 CoDA ZFNs function effectively in vivo in zebrafish, generating indel mutations at a detectable frequency (1.1%) in injected embryos. Notably, we have achieved variable success with CoDA ZFN array construction, as only one (prp1) out of the four (nlz2, sfrp5, and gdf11) CoDA ZFNs that we tested successfully cleaves target-site DNA in vitro (data not shown).
Not all ZFNs constructed through context-dependent selection strategies function effectively to generate somatic mutations. Testing ZFNs in vivo can be time consuming and expensive, especially in instances where mutation frequencies are quite low. We, therefore, elaborated on published work 5 and established a system to rapidly gauge the target-site-specific cleavage activity of ZFNs in vitro. We demonstrate that this system can be used to evaluate the target-site-specific cleavage activity of ZFN pairs. In all cases examined, those ZFNs that functioned to cleave target-site DNA in our in vitro assay also generated somatic mutations in vivo in zebrafish. Our results, therefore, establish our in vitro cleavage assay as a good predictor of ZFN fitness in in vivo mutagenesis applications. Notably, in addition to evaluating ZFN function, we demonstrate that our in vitro cleavage assay can also be used to evaluate the target-site-specific cleavage activity of other synthetic restriction endonucleases, such as TALENs.
The mutagenesis efficiency of a ZFN is partially dependent on the catalytic activity of its endonuclease domain. 107 We enhanced the catalytic activity of our ZFNs using the Sharkey FokI endonuclease domain variant. We demonstrate that Sharkey ZFNs exhibit increased in vitro cleavage of target-site DNA compared with control ZFNs. Our results also suggest that less Sharkey ZFN protein is required to elicit site-specific DNA cleavage in vitro. More importantly, we demonstrate that, in limited instances, Sharkey ZFNs have the capacity to exhibit greater in vivo mutagenic activity than control ZFNs, producing approximately a 26-fold increase in the indel mutation frequency of injected zebrafish embryos (Table 1). This expands on previous research in cell culture, which has shown that Sharkey ZFNs demonstrate three- to six-fold more mutagenic activity in HEK 293 cells when compared with wild-type ZFNs. 107 Notably, ours is the first study that assesses the relative mutagenic activity of control and Sharkey ZFNs in vivo in embryos. We did not systematically evaluate the off-target cleavage activity of Sharkey ZFNs in zebrafish. However, in comparison to control ZFNs, we find that Sharkey ZFNs do not increase the frequency of morphological defects, or the mortality of injected embryos. Our overall results suggest that incorporating the Sharkey FokI endonuclease domain into ZFNs may be a simple method of enhancing their mutagenic activity in vivo. Notably, introducing double-strand DNA breaks near a desired recombination site can dramatically increase the frequency of homologous recombination in mammalian cells.122,123 ZFNs have previously been used in this capacity.8,10 Consequently, the enhanced catalytic activity of Sharkey ZFNs may also be extremely beneficial for homologous recombination-mediated genome engineering applications.
TALENs have recently been shown to be approximately 10 times more mutagenic than ZFNs in zebrafish. 106 Furthermore, TALENs can be targeted to nearly any DNA sequence, and their modular nature makes them easy to design and assemble. For these reasons, TALENs are quickly becoming the technology of choice for targeted mutagenesis in animal models. Notably, TALENs have recently been used to elicit genome modification by homologous recombination, albeit at low frequency.95,124
Given the partial success that we achieved in increasing the mutagenic activity of our ZFNs using the Sharkey FokI endonuclease variant, we sought to determine whether this variant could also be applied to TALENs to enhance their catalytic activity. We demonstrate that Sharkey TALENs exhibit increased in vitro cleavage of target-site specific DNA compared with control TALENs. However, unlike with ZFNs, we demonstrate that Sharkey TALENs exhibit significantly reduced in vivo mutagenic activity in injected zebrafish embryos when compared with control TALENs (Table 2). Our overall results suggest that incorporating the Sharkey FokI endonuclease domain into TALENs may severely abrogate their in vivo mutagenesis function in zebrafish.
Footnotes
Acknowledgments
The authors thank members of the Univeristy of Alberta ZFN consortium, Scot Wolfe, and J. Keith Joung for technical assistance and constructive comments. They also thank Timothy Dahlem for advice regarding HRMA; Aleah McCorry and Erin Wilson for fish care.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.