
Abstract
Abstract
We evaluated the use of the gnotobiotic zebrafish system to study the effects of bacterial infection, and analyzed expression of genes involved in zebrafish innate immunity. Using a GFP-labeled strain of Vibrio anguillarum, we fluorescently monitored colonization of the zebrafish intestinal tract and used gene expression analysis to compare changes in genes involved in innate immunity between nongnotobiotic and gnotobiotic larvae. The experiments performed with the gnotobiotic zebrafish reveal new insights into V. anguillarum pathogenesis. Specifically, an alteration of the host immune system was detected through the suppression of a number of innate immune genes (NFKB, IL1B, TLR4, MPX, and TRF) during the first 3 h post infection. This immunomodulation can be indicative of a “stealth mechanism” of mucus invasion in which the pathogen found a sheltered niche, a typical trait of intracellular pathogens.
Introduction
M
The innate immune system of fish acts as the first line of host defense against microbial pathogens by detecting and responding to a broad range of invading pathogens directly after infection. 12 The immune response of fish against VAN infection is largely unknown. 13 Previous studies have revealed than VAN infection inhibits the respiratory burst of rainbow trout macrophages 14 and sea bass leukocytes, allowing the bacteria to survive inside phagocytes. 8 Rojo et al. described a rapid induction of defence genes within 2 hpi with VAN in zebrafish adults, characterized by a steady increase of expression starting from the initial stages of the interaction. 15 Zebrafish is a powerful vertebrate model organism for immunological research,16,17 as this model has been extensively used to study the host immune response under a number of microbial infections5,9,15,18–24 as well as the interactions between the host and the natural gut microbiota.25–32
The aim of this work was to better understand the mechanism of action of VAN pathogenicity. Our hypothesis was that the use of gnotobiotic zebrafish larvae would permit us to test the innate immune response of zebrafish to VAN infection, thus allowing us to better understand the mechanism of VAN pathogenicity. To test our hypothesis, we first monitored VAN colonization of the zebrafish intestinal tract by employing a GFP-labeled strain of VAN bacteria; subsequently, we evaluated expression of a selected group of genes and compared the expression profile of these genes between gnotobiotic and nongnotobiotic larvae.
The set-up of this experimental system will enable the analysis of temporal changes in host-pathogen interactions, including pathogenicity and host immune response. It will also enable the investigation of the effects of different microbial communities on host immunology and host nutrition, as well as the study of microbial composition and activity, such as the potential beneficial effect of probiotics.
Materials and Methods
Zebrafish husbandry
Adult zebrafish (Danio rerio, Hamilton 1822) were maintained at 27°C in 60 l tanks, with aerated freshwater. Each tank contained an external filtration system (biological, chemical, and physical filtration), supplemented by an ultraviolet lamp. Zebrafish were maintained according to standard protocols. 33
Fish were fed with a pellet-formulated diet (Gemma Micro 300; Skretting) to achieve a total daily feed input of 5% of body weight per day and were reared on a 12-h light/12-h dark cycle.
Protocol for obtaining gnotobiotic larvae
Disinfection of zebrafish embryos
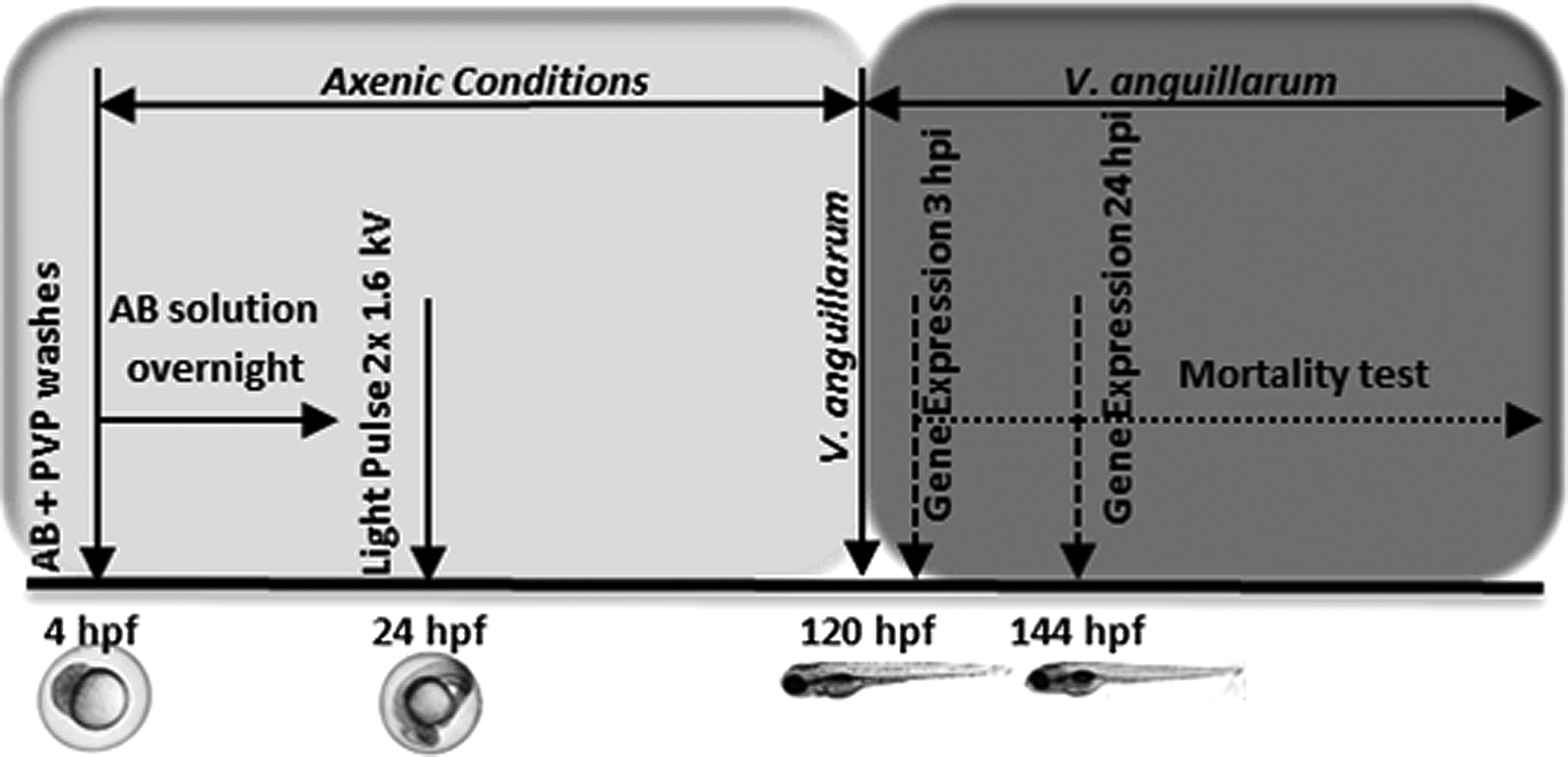
Embryos were collected directly from the breeding tanks immediately after fertilization. After egg collection, the entire procedure was set under a laminar flow cabinet to maintain sterile conditions. Sterile solutions and materials were also used. The fertilized embryos were washed with a sterilized EWB solution [Embryo water (EW): CaCl2 294 mg/mL, MgSO47H2O 123.3 mg/mL, NaHCO3 63 mg/mL, and KCl 5.5 mg/mL, supplemented with methylene blue 0,01% (w/v)], to remove the fecal matter. Embryos were carefully collected with a sterile disposable plastic pipette, transferred to a 15 mL sterilized conical tube, and washed ten times in EWB. Embryos with visible debris attached to the chorion were eliminated. 34
After the initial wash step, embryos were next washed ten times in an antibiotic solution (AB solution). The AB solution includes a pool of three antibiotics: kanamycin (15 μg/mL,), ampicillin (300 μg/mL), and amphotericin b (1.25 μg/mL) with antifungal activity. The concentration and variety of antibiotics used is a crucial factor. We modified the protocol previously described by Pham et al. (2008) 34 by increasing final antibiotic concentrations. After the antibiotic wash, the AB solution was removed and embryos were gently immersed in 0.02% (w/v) Polyvinylpyrrolidone (PVP) for 2 min. The PVP excess was immediately removed by washing the embryos ten times in EWB. The timing and final concentration of the PVP treatment should be strictly controlled, because the PVP solution is extremely toxic to aquatic life. After PVP treatment, embryos were incubated in a 0.003% (v/v) bleach solution for 1 h, and subsequently washed ten times in sterile EWB solution. Dead embryos were removed to minimize the growth of contaminating microorganisms, and embryos were incubated overnight in AB solution.
The next day, the AB solution was removed by washing the embryos ten times in sterile EWB solution. Fifty embryos were collected and transferred to a Petri plate (5.5 cm diameter×1.0 cm) containing 5 mL EWB solution, and treated with two UV-light pulses of 1.6 kV using a Pulsed Light equipment (Pulsed UV System XeMatica 1:2L-SA, SteriBeam Systems, GmbH) to inactivate the microbial burden present in the sample.
Figure 1 describes the procedure schematically. The gnotobiotic embryos obtained were raised under axenic conditions during the first 6 days post fecundation (dpf).

Schematic overview of the procedure for obtaining gnotobiotic zebrafish.
Effect of UV light pulses on larvae survival
To identify potential side-effects of UV light pulses on larvae, embryos were monitored at 5 and 6 dpf for defects such as malformation, hatching delays, and mortality. Abnormal embryos were discarded. At 6 dpf, gnotobiotic zebrafish larvae were transferred into tanks with circulating, filtered water flow and grown until adulthood (6 months). To determine the potential side-effects of UV light pulses on later stages of the development, larvae were periodically examined by visual inspection under a Leica MZFL III stereomicroscope. Malformations and developmental delays were determined by visual inspection; mortality was identified by larvae coagulation or by lack of a heartbeat. Adult fish fertility was evaluated by checking the number and viability of laid embryos.
Axenity test
Axenity was tested after the UV light pulse treatment, when larvae were 96 h post fecundation (hpf). The evaluation of sterility from the gnotobiotic zebrafish embryos was tested by (i) the culturing of 1 mL of water and (ii) by culturing a pool of fifteen 3–6 dpf zebrafish larvae after homogenization with a Pellet Pestle Cordless Motor (Kimble Chase Life Science and Research Products LLC) in 1 mL of EWB. In both cases, samples were cultured under aerobic and anaerobic conditions on a nonselective microbiological growth medium Plate Count Agar (PCA) and brain/heart infusion broth (BHI) at 30°C during 48–72 h to monitor total and viable bacterial growth. Samples were also cultured on 10 mL of Sabouraud Dextrose Broth (Sab-Dex) and incubated at 25°C for 3–5 days to detect the presence of yeasts, molds, and aciduric microorganisms. In addition, a pool of five larvae was used as template for PCR amplification using primers targeting 16S ribosomal RNA gene to determine the presence of any bacteria inside larvae. 26 Larvae were solubilized in 0.5 mL of an extraction buffer [1% (w/v) SDS, 150 mM NaCl, 2 mM EDTA, 10 mM Tris-HCl, pH 8.0, supplemented with 50 μL of 5 M guanidinium thiocyanate, and 25 μL of proteinase K solution (Applied Biosystems)]. The mixture was incubated at 56°C overnight and then centrifuged at 8000 g for 5 min. The supernatant was treated with the Wizard-DNA Clean-Up Extraction Kit (Promega). The purified DNA was resuspended in bidistilled sterile water and stored at −20°C. Quantitative PCR was carried out with a Light Cycler 480 sequence detection system (Roche Diagnostics) in a 10 μL solution containing 300 nM primers, 5 μL 2x SYBR Green PCR master mix (Roche Diagnostics), and 40–50 ng of DNA template. For routine sterility tests, water was cultured in PCA, BHI, and Sab-Dex plates under aerobic conditions.
V. anguillarum culture conditions and infection procedure
V. anguillarum serotype O2a, strainsNB10 GFP- labeled, 5 kindly provided by R. O'Toole and H. Wolf- Watz from Umeå University, was tested for its pathogenic effect in zebrafish larvae. The bacteria were grown at 25°C in TSB (trypticase soy broth) to logarithmic growth phase (16 h). An antibiotic concentration of 10 μg/mL chloramphenicol and 0.5 mM IPTG (isopropyl-β-D-thiogalactopyranoside) was used as described by O'Toole et al. (2004). Cells were spun down (1.150 g at 25°C for 10 min), washed twice with sterilized embryo water with a salinity of 5 ‰ (EW5), and re-suspended in EW5 at the desired density.
Determination of colony-forming units (CFU) was performed in duplicate by spreading 100 μL of resuspended bacteria on trypticase soy agar with chloramphenicol.
In a preliminary experiment, the effect of salinity on survival rate of the bacteria was analyzed. No decrease in CFUs/mL was detected after an incubation of 72 h for bacteria resuspended in EW5.
Challenge test
Groups of 20 gnotobiotic and nongnotobiotic larvae at 120 hpf were immersed in a final concentration of VAN at 108 CFUs/mL. These two groups were designated NGV and GV, respectively. Control groups were immersed in EW5. Mortality, malformations, and developmental delays were determined at 3, 24, 48, and 72 hpi (Fig. 2).
Scheme of gnotobiotic larvae infection.
Detection of bacteria inside the fish larvae
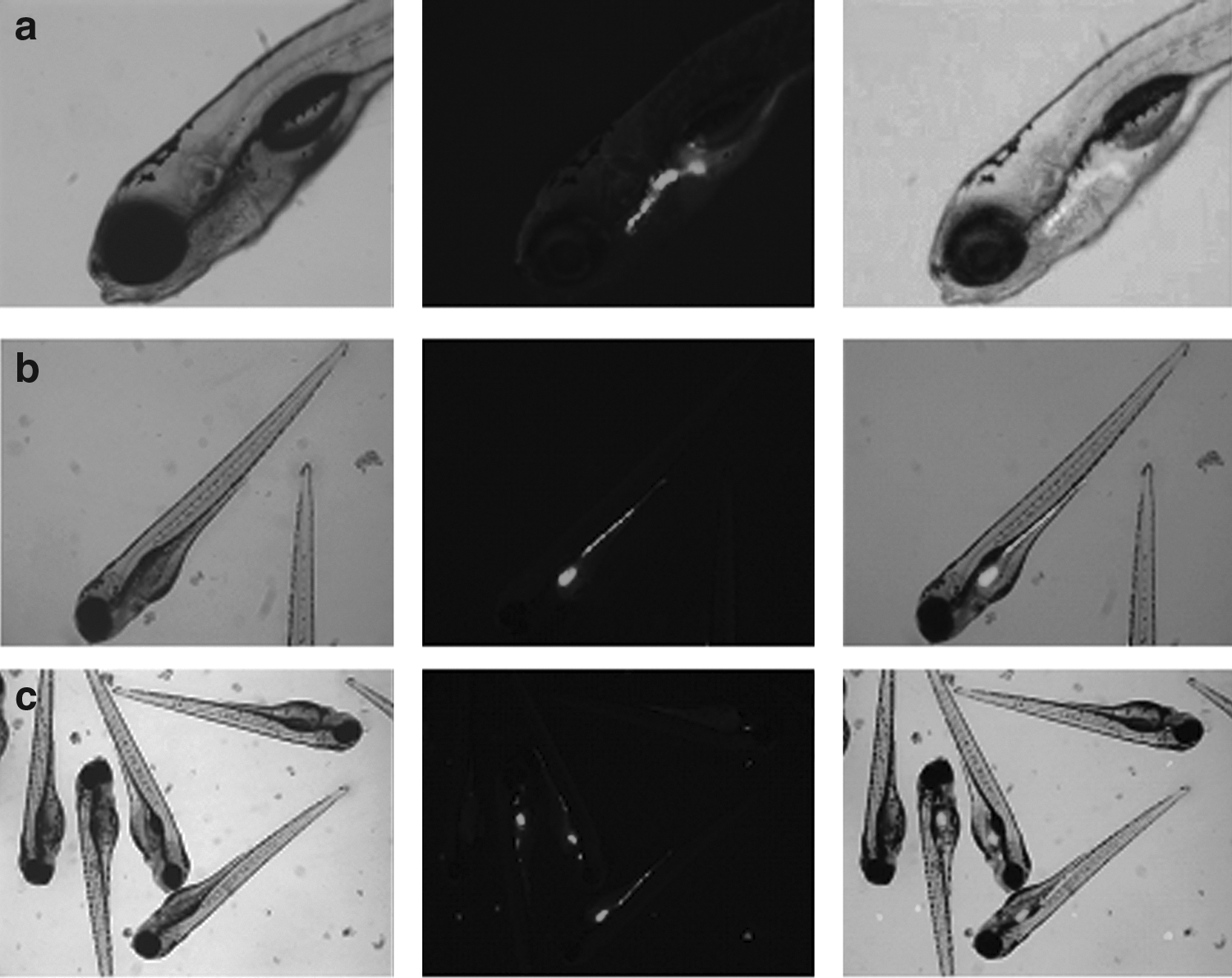
The colonization of bacteria inside the larvae was assessed visually using a Leica MZFL III stereomicroscope with a zoom magnification range of 8×to 100×. The microscope was equipped with visible light and UV light (Hg 100 W) sources. GFP fluorescence was detected by exposure of the larvae to UV light in the excitation range of 450–490 nm. Images were captured using a Leica DFC 360FX camera and processed using ImageJ software v1.47 (National Institutes of Health, NIH) to obtain the integrated density value (the product of area and mean fluorescence value). Three pools of five infected larvae at 3 and 24 hpi were homogenized with a pestle and cultured, as previously described in V. anguillarum culture conditions and infection procedure section, to count the number of CFU inside the larvae.
Mucus secretion evaluation
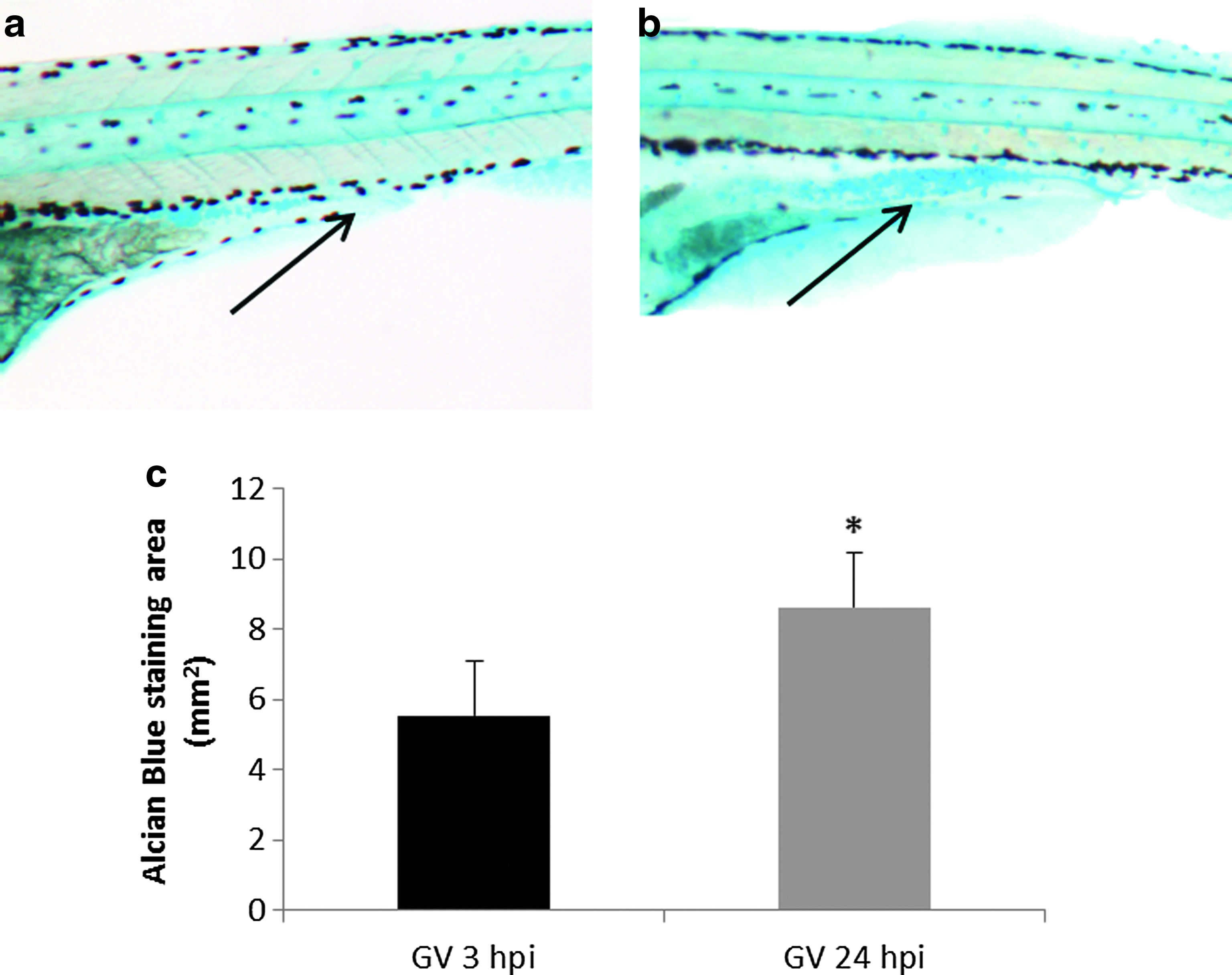
Using Alcian blue staining, mucus secretion in whole-mount larvae was visualized with a Nikon SMZ1000 stereomicroscope as previously described by Chen et al. 35 Fifteen larvae per condition, in two independent experiments, were automatically analyzed and processed to obtain staining area (mm2) through the “Colour Deconvolution” plug of ImageJ software v1.47 (National Institutes of Health, NIH). 36 The p-values were calculated with the Statgraphics software v16.1.17 (StatPoint Technologies, Inc.), and differences were considered statistically significant at p<0.05.
Gene expression analysis (RT-qPCR)
To study gene expression, groups of 10–15 larvae were sampled at 3 and 24 hpi. Three replicates per group were analyzed in three independent experiments.
The quantity and quality of RNA samples were determined by capillary electrophoresis using an Agilent 2100 Bioanalyzer (Agilent Technologies). The Bioanalyzer provides an RNA integrity number (RIN), with 0 corresponding to fully degraded RNA and 10 corresponding to intact RNA. 37 In all experiments, only RNA samples with a RIN of at least 8.5 were used. These values fulfil one of the requirements of a satisfactory qPCR experiment. 38 cDNAs were synthesized from the RNA samples in a reverse transcription reaction (RT) containing 40 ng of RNA per assay. RT was performed in a mix containing 1x TaqMan RT buffer, 5.5 mM MgCl2, 500 μM dNTPs, 2.5 μM oligo-dT, RNase inhibitor (0.4 U/μL), and 1.25 U/μL MultiScribe reverse transcriptase (Applied Biosystems). The mixture was incubated at 25°C for 10 min and at 48°C for 30 min, and the enzyme was inactivated at 95°C for 10 min.
Changes in mRNA expression of genes related to the innate immune system were monitored using real-time qPCR. The following genes were analyzed: interleukin 1β (IL1β), transferrin (TRF), myeloperoxidase (MPO), lysozyme (LYZ), toll like receptor 4 (TLR4), toll-like receptor 22 (TLR22), and nuclear factor κ β (NFKB). Primer sequences are listed in Table 1.
Quantitative PCR was carried out with a Light Cycler 480 sequence detection system (Roche Diagnostics). Each reaction was performed in a 10 μL solution containing 300 nM primers, 5 μL 2x SYBR Green PCR master mix (Roche Diagnostics), and 10 ng of cDNA template.
Reaction conditions were as follows: 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. A dissociation step was performed at the end of the PCR: 95°C for 15 s, 60°C for 20 s, and 95°C for 15 s.
Each PCR reaction was performed in triplicate, and β-actin was used as a housekeeping gene to normalize the samples The threshold cycles and copy number for each reaction were calculated by the Light Cycler 480; RNA expression levels were calculated by the 2−ΔΔCt method. 39 Results were expressed as a mean±standard error. The p-values were calculated using the software REST2009 (www.REST.de.com) (Qiagen). Differences were considered statistically significant at p<0.05.
Results
Generation of gnotobiotic zebrafish larvae
To ensure that our protocol for obtaining gnotobiotic larvae was effective in removing all microorganisms from the larvae, we homogenized 10 pools of 15 larvae at 3 and 6 dpf and incubated under aerobic and anaerobic conditions in PCA, BHI, and Sab-Dex plates. In addition, ten pools of five larvae were tested by PCR amplification using primers targeting of 16S ribosomal RNA genes to check the absence of any bacterial contamination inside larvae. Using the protocol described here, we have typically obtained 90%–95% sterility rates through 3–6 dpf. In any case, if a particular sample was contaminated those fish were immediately removed from the experiment. The homogenized larvae samples, which had undergone antibiotic and UV light pulse treatments, did not show microbial growth on the PCA, BHI, and Sab-Dex plates after incubation or PCR amplification compared with nontemplate controls.
Potential negative side-effects of the disinfection process were determined by studying the survival rates and presence of malformations in the treated larvae, as well as the fertility in the corresponding adults. Survival rates were evaluated at 5 days after treatment with antibiotic and UV light pulse to study any possible negative effects on larvae development. The survival rate in the treated larvae was slightly lower than that in the control group. The deformity rate was 5.5% in the treated larvae, while we did not find malformations in the control group. However, in both cases, differences were not statistically significant.
To ensure that the treatment had no effect on the normal development of the treated larvae, gnotobiotic larvae were conventionalized at 5 dpf with tank water and maintained under normal conditions. After 6 months post fertilization, no deformities were found in treated or control groups. Survival rates were slightly higher in the control group than in the treated group; however, no significant differences were found between them. Moreover, the treated group was able to mate and produce viable embryos.
V. anguillarum challenge test
Bacteria entered through the larvae mouth; after 3 hpi, they were detected mainly in the first part of the digestive tract (Fig. 3a) and after 24 hpi throughout the digestive tract (Fig. 3b). Gut colonization was highly variable between larvae in each sample both at 3 hpi and after 24 hpi. Bacteria were detected throughout the entire digestive tract in some larvae, while in others; colonization was limited to specific sections (Fig. 3c). However, after obtaining media of fluorescent integrated density, the highest percentage of VAN was quantified in the anterior section of the intestine at 3 hpi and in the mid/posterior section at 24 hpi (Fig. 4). At 3 hpi, the total fluorescent area detected was 3.76 (±1.08)*103 mm2/larva and the bacterial load 7.8 (±1.17)*105 CFU/larva. At 24 hpi, the values detected were 4.40 (±0.73)*103 mm2/larva and 6.2 (±1.4)*105 CFU/larva.
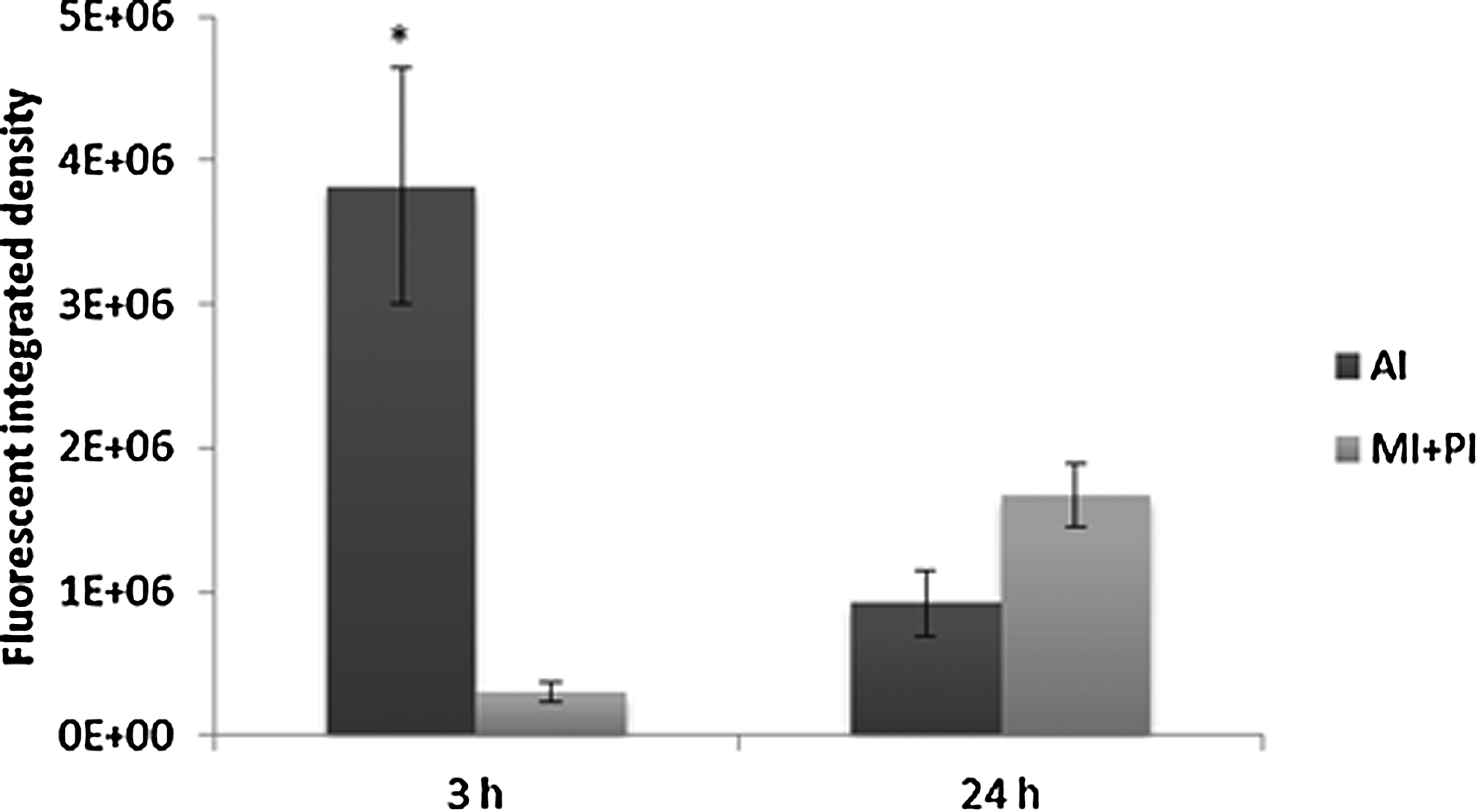
Bacteria quantification after 3 and 24 hpi in anterior intestine (AI) and mid/posterior intestine (MI+PI). Percentages indicate the fluorescent integrated density media detected throughout the intestine of 10 representative larvae. Error bars indicate the standard error of the mean. Differences between 3 and 24 hpi were considered statistically significant at p<0.05 (*).
The mortality rate of different VAN exposures was variable after 24 hpi and ranged from 5% to 60% (data not shown). Using a stereomicroscope to monitor VAN colonization, it was found that the presence and amount of VAN in GV and NGV larvae was the same (data not shown). No significant differences in mortality were detected between the two groups after 24 and 48 hpi; the mortality rate was 100% for both groups after 72 hpi.
Immune response induced on V. anguillarum infection
Mucus secretion was visualized using Alcian blue staining on whole-mount gnotobiotic and nongnotobiotic larvae, with and without VAN inoculum, at 3 and 24 hpi. A statistically significant increase in mucus production was detected in gnotobiotic larvae after 24 hpi with VAN when compared with 3 hpi with VAN. (Fig. 5). The mucus was primarily located in the mid-intestine of the larvae. No differences were detected between gnotobiotic and gnotobiotic with VAN (GV) larvae at either 3 or 24 hpi (data not shown).
Seven key genes directly related to the innate immune cascade were investigated using gene expression analysis on VAN infection. Three independent experiments were carried out, and all samples were tested in triplicate. Six of the seven innate immune genes analyzed showed a “suppression profile” in the gnotobiotic group. After 3 hpi with VAN, genes NFKB, TLR4, and TRF were consistently downregulated in the gnotobiotic group throughout all experiments. Genes IL1B, MPO, and TLR22 were also downregulated in the gnotobiotic group but results were not consistent between experiments. Lysozyme (LYZ) did not show any change in gene expression between groups. Conversely, the IL1B gene was steadily upregulated in the NGV group, in all experiments performed. The remaining genes showed high variability in expression between experiments in the NGV group. (Fig. 6).

Gene expression analysis after 3 and 24 h on VAN infection (hpi) in gnotobiotic (GV) and nongnotobiotic (NGV) larvae in three independent experiments (highlighted with different gray gradients). Expression differences were obtained after normalization with the housekeeping gene and a comparison with “no infected with VAN” control samples. Each experiment represents the mean of three replicates, and error bars indicate the standard error of the mean. Differences were considered statistically significant at p<0.05 (*) and p<0.01 (**).
After 24 hpi, most genes tested were upregulated in both the GV and NGV groups. The GV group showed a clear upregulation in five of the seven genes analyzed: IL1B, LYZ, MPO, TLR4, and TLR22; NFKB showed the same enhanced expression profile in two of the three triplicates analyzed. Similar results were achieved in the NGV group (Fig. 6). While gene expression variability was observed in all groups and time points tested, the gnotobiotic group showed a greater repeatability within experiments at both times analyzed (3 and 24 hpi).
Discussion
The objective of this work was to use gnotobiotic zebrafish larvae as a model to study the mechanism of action of V. anguillarum pathogenicity. To reach this aim, we developed a novel and efficient protocol employing a combination of antibiotics and pulsed UV light to produce gnotobiotic embryos.
The protocol used to obtain gnotobiotic zebrafish embryos was based on the method previously published by Pham et al., 2008, with a revision to include natural breeding to eliminate any stressful fish procedure and abide by the 3Rs principles. Unlike other procedures used to obtain gnotobiotic zebrafish embryos, such as squeezing and laparotomy, natural breeding does not affect fish welfare. 34 However, one main disadvantage of natural breeding is the relatively higher initial microbial burden. To overcome this problem, we employed a novel sanitization technology: denominated pulsed light. This treatment enhances the antimicrobial effects produced by the combination of antibiotics, PVP, and bleach washing. The pulsed light technology is based on the ability of UV and visible light to inactivate cellular microorganisms and viruses by short duration pulses (a few hundreds of milliseconds) of broad spectrum wavelengths (from UV to near infrared) on the surface of materials and in transparent media such as water.40–44 This combination of UV wavelengths with visible and infrared regions kills microorganisms such as bacteria, yeast, and fungi. To our knowledge, this is the first time that pulsed light technology has been used to obtain gnotobiotic aquatic animals. Therefore, it is very important to evaluate the effect of this new technology on the development of zebrafish larvae. We found that our modified protocol did not significantly affect survival rates of zebrafish larvae, which was consistent with previously reported results. 34 In addition, our protocol did not compromise the sterility of gnotobiotic larvae; as a matter of fact, slightly lower sterility rates were reported by other authors. 34 Since we did not detect the growth of any anaerobic bacteria, for routine sterility tests we have eventually chosen the cultivation of EWB under aerobic conditions. Moreover, facultative anaerobes will be also detected under aerobic conditions and the presence of strict anaerobes in aerobic aquatic environment is conditioned by the previous presence of facultative anaerobes in larvae gut. 32
Negative effects as a result of pulsed light exposure in axenic conditions were not observed during the first 5 days of development. In addition, no significant differences in mortality or deformity rates were observed between treated and control groups grown in normal conditions, with nonsterile tank water, to 6 months of development. This indicates that the disinfection procedure, the number and intensity of pulsed light, is strong enough to destroy microorganisms without affecting the normal development of larvae and fish.
Although numerous papers have described bacterial pathogenesis in fish, to our knowledge, none have used gnotobiotic zebrafish larvae to investigate the pathogenesis and mechanism of action of any bacteria, including VAN. Gnotobiotic fish larvae provide a very useful tool to study host-pathogen interactions because the removal of stochastic and nuisance bacterial colonization contributes to improve repeatability and reproducibility. 9 We observed a higher reproducibility of results in the gnotobiotic group compared with the nongnotobiotic group. The variability we observed within and between experiments may be caused by the fact that the larvae were present in a pool originated from different hatchings and different parents. In previous studies of zebrafish infection by immersion with VAN, a high variability in larvae mortality was found during the first 4 days after infection when comparing experiments carried out using larvae from different hatchings. 45 Differences in hatchings might result in differences in developmental staging, resulting in variations in timing of mouth opening and bacterial colonization. Gene expression variability was higher after 3 hpi than after 24 hpi in both groups. This may be due to an initial variation in bacterial concentrations within the larvae and a shorter exposure time, which may, in turn, influence mechanisms of gene activation. Other studies with Edwarsiella tarda also found important variability in mortality rates after immersion with bacteria. 46 The variability observed in larvae mortality and gene expression may also be due to the fact that not all embryos become systemically infected using this method, limiting analysis to those larvae that have an innate immune response to the presence of bacteria. However, we were investigating changes in gene expression as a general response to bacterial infection independent of hatching; we found that gnotobiotic larvae gave a more homogenous response to bacterial infection than nongnotobiotic larvae, and displayed a higher reproducibility of results than nongnotobiotic larvae. The consistent results seen within the gnotobiotic larvae group were most likely due to the absence of other bacteria in the gnotobiotic larvae.
In this study, we have used VAN, a pathogen that can enter fish cells and survive as intracellular bacteria.2,8 The success of a pathogen depends on its ability to overcome the innate and adaptive immune response within the host. Some bacteria have different evasion strategies to escape from the host immune response. 47 Human microbial pathogens, such as Fransciella tularensis, can evade these defenses by inhibition of the respiratory burst. 48 In a recent investigation, the pathogenic colonization of gut enterocytes after 2 h post VAN exposure was described in gnotobiotic sea bass larvae. 7 In addition, some studies have detailed the ability of VAN to survive intracellularly in epithelial cell lines, 2 gut enterocytes, 7 and macrophages and to inhibit the respiratory burst of leukocytes, 8 suggesting that some VAN strains are intracellular pathogens. 14 However, none of these investigations provide evidence of the molecular mechanism action of VAN in the host without the interference of the natural microbiota, and are rather investigations into the virulence mechanisms of intracellular pathogens. Another likely virulence mechanism of intracellular pathogens is the “stealth mechanism,” the suppression of NFKB signal transmission to elude the host immune system. 49 Our results in the gnotobiotic system showed a clear suppression of NFKB, after 3 hpi, along with suppression of other markers directly involved in the immune response and pro-inflammatory cascade, such as IL1B, MPO, TLR4, TLR22, and TRF. The downregulation of these genes suggests that VAN may elude the larva's innate defense mechanisms as a “stealth mechanism” during the first stages of pathogen invasion. However, we were not able to detect this suppression effect in the nongnotobiotic system, probably due to the presence of natural microbiota that may mask the mechanisms of action of VAN pathogenicity. Therefore, removing indigenous microbiota, such as in gnotobiotic zebrafish, may provide an excellent tool in understanding the modes of action of host-microbe interactions.7,34
After 24 h post VAN infection (hpi), the progression of pathogens throughout the intestine produced a generalized activation of the majority of genes analyzed in this study, including upregulation of IL1B, a marker of inflammation in zebrafish. 15 This inflammatory effect produced an activation response that was seen in both gnotobiotic and nongnotobiotic systems. After 24 hpi, mucus secretion, a defense mechanism of the host larvae, was detected and larvae started to die. After 96 hpi, mortality reached 100% in both systems.
In this study, we demonstrate that the use of gnotobiotic fish larvae has considerable advantages in early detection of an infection and provides helpful insights into the mechanism of action of a given pathogenic microorganism. The results obtained in this study lead us to speculate a “stealth mechanism” as a part of virulence behavior of VAN to elude the defense system of the host. However, it must be taken into account that real-life conditions may be far more complex and any findings made under gnotobiotic conditions will need to be validated in more definitive conditions. 9 As such, this new insight in VAN virulence should be complemented in future works in gnotobiotic and nongnotobiotic organisms. However, this does not diminish the importance of the findings detailed in this study. The zebrafish gnotobiotic model is a useful tool to study host–microbial interactions, as it allows for the evaluation of early stages of infection and provides an opportunity for increased repeatability and reproducibility of host-microbial interactions.
Footnotes
Acknowledgments
The authors sincerely thank R. O'Toole and H. Wolf- Watz (Umeå University, Sweden) for providing them with the GFP-marked V. anguillarum strain and Rebecca Anderson (Northwestern University, USA) for improving the English language of this article. This study was financially supported by the Agriculture and Fisheries Department of the Basque government (project IA2009Daretest), and UO is the recipient of a PhD fellowship from Iñaki Goenaga Foundation.
This article is the contribution of n 696 from AZTI-Tecnalia (Food Research Division).
Disclosure Statement
No competing financial interests exist.