
Abstract
Abstract
Testicular tumors are the most common solid malignant tumors in men 20–35 years of age. Although most of testicular tumors are curable, current treatments still fail in 15%–20% of patients. However, insufficient understanding of the molecular basis and lack of animal models limit development of more effective treatments. This study reports the identification of a novel zebrafish mutant line, ns1402, which develops testicular germ cell tumors (TGCTs). While both male and female ns1402 mutants were fertile at young age, male ns1402 mutants became infertile as early as 9 months of age. This infertility was associated with progressive loss of mature sperm. Failure of spermatogenesis was, at least in part, explained by progressive loss of mature Leydig cells, a source of testosterone that is essential for spermatogenesis. Interestingly, TGCTs in ns1402 mutants contained a large number of Sertoli cells and gene expression profiles of Sertoli cells were altered before loss of mature Leydig cells. This suggests that changes in Sertoli cell properties happened first, followed by loss of mature Leydig cells and failure of spermatogenesis. Taken together, this study emphasizes the importance of cell–cell interactions and cell signaling in the testis for spermatogenesis and tissue homeostasis.
Introduction
Testicular tumors are the most common solid malignant tumors in men 20–35 years of age. Consequences of testicular tumors are magnified because it mostly strikes males who are young men of childbearing age. While most of testicular tumors are solid, 10%–20% of these tumors metastasize and show resistance to chemotherapy or radiation.1,2 Further understanding the mechanisms underlying testicular tumor formation is essential to develop more effective therapeutic protocols.
The zebrafish is a valuable model organism to study potential genetic and environmental risk factors that may influence tumor formation.3–7 Because of the organism's small size, it is possible to analyze tumor formation in a large number of animals with multiple genetic backgrounds, environments, and age. Previous studies have reported mutant or transgenic zebrafish lines that developed testicular germ cell tumors (TGCTs). For instance, zebrafish mutants of alk6b, a gene encoding a receptor protein for bone morphogenetic protein (BMP), developed TGCTs. 8 These fish showed reduced activation of BMP signaling in TGCTs, which has also been reported in human TGCTs. 9 Mutations in lrrc50, a gene encoding a component of the basal body/centriole complex, were also identified both in zebrafish and human TGCTs. 10 These studies suggested that molecular mechanisms underlying TGCT formation might be largely conserved among vertebrates and that zebrafish could be used as a model to study molecular mechanisms underlying human TGCTs.
In this study, we report identification of ns1402, a new zebrafish mutant line that develops TGCTs. In contrast to the majority of human TGCTs that contain only spermatogonial germ cells, TGCTs in ns1402 mutants contained a large number of immature Sertoli cells, in addition to undifferentiated spermatogonial germ cells. While both female and male ns1402 mutant zebrafish were fertile at young ages, male ns1402 mutants became infertile as young as 9 months of age. The failure of spermatogenesis was, at least in part, explained by loss of mature Leydig cells, a source of testosterone that is an essential androgen for proceeding meiosis. Changes in gene expression of Sertoli cell-specific markers were observed before loss of mature Leydig cell makers. Since Sertoli cells produce factors required for the maturation of Leydig cells, these results suggest that Sertoli cells progressively failed to produce mature Leydig cells, resulting in impaired spermatogenesis in ns1402 mutants. Taken together, these findings emphasize the importance of cell–cell interactions and cell signaling in the testis to promote spermatogenesis and maintain tissue homeostasis.
Materials and Methods
Zebrafish maintenance
Zebrafish were maintained under standard conditions as described previously. 11 The use of animals was approved by the Institutional Animal Care and Use Committee at Rutgers University. Control AB wild-type fish were purchased from the Zebrafish International Resource Center. The breeding strategy of ns1402 mutants was as follows: prospective ns1402 mutant fish <9 months of age were used for breeding. It should be noted that the majority of prospective ns1402 mutants did not show significant changes in their gross morphology or fertility at that age. Therefore, the inheritance of the ns1402 mutation had not been determined at the time of breeding. At 1.2–1.5 years of age, inheritance of the ns1402 mutation was determined by the development of TGCTs in the fish used for breeding and their male siblings. Only progenies of zebrafish in which inheritance of ns1402 mutation was confirmed were raised and used for further analysis.
Dissection of testis and hematoxylin and eosin staining
Adult zebrafish were euthanized by overdose of tricaine-S methanesulfonate (Western Chemical, Inc.) before dissection. Paraffin embedding and hematoxylin and eosin (H&E) staining were performed by the Rutgers Confocal Imaging facility using standard protocols. Briefly, the isolated testes were fixed in modified Davidson's fluid, containing 10% formaldehyde and 5% glacial acid overnight at 4°C. 12 The testes were then embedded in paraffin and sectioned at 6 μm using a microtome (Leica). After H&E staining, tissue sections were mounted on Fisher Scientific Premium Glass Microscope Slides and images were taken by the Olympus BX51 microscope equipped with a color CCD camera (DP72; Olympus).
Immunohistochemistry
The testes were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in phosphate-buffered saline (PBS) overnight at 4°C, followed by overnight incubation in 30% sucrose in PBS. The testes were then embedded in Tissue-Tek OCT compound (Sakura, Japan) and sectioned at 6 μm thickness using a cryostat microtome (Leica). After incubation in blocking buffer (10% normal goat serum in PBS), cryosectioned samples were immunostained with an anti-Sox9 mouse monoclonal antibody (Abcam; clone 3C10) diluted 1:500. After washing in PBS 3 times, an Alexa Fluor 488 conjugated goat anti-mouse immunoglobulin G antibody (Invitrogen) was applied. 4′,6-Diamidino-2-phenylindole (DAPI) (Invitrogen) diluted 1:1000 or Alexa Fluor 543 phalloidin (Invitrogen) diluted 1:500 was used to counterstain nuclei or actin filaments. After mounting on glass microscope slides, images were taken by the Olympus BX51 microscope equipped with a color CCD camera (DP72; Olympus). ImageJ software with the Cell Count plugin was used to count the number of Sox9-positive nuclei in the entire image areas.
RNA isolation and real-time PCR
Total RNA was isolated using TRIzol Reagent following the manufacturer's guideline (Invitrogen). iScript Reverse Transcription Supermix (Bio-Rad) was used for reverse transcription. Quantitative real-time PCR was conducted using the Power SYBR Green PCR Master Mix (Applied Biosystems) and Applied Biosystems 7500 Real-time PCR system. Primers used for real-time PCR are listed in Table 1.
List of Primers Used in Reverse Transcription-Quantitative Polymerase Chain Reaction Analysis
Whole-mount in situ hybridization
In situ hybridization was performed as described previously. 13 To generate DIG-labeled antisense RNA probes, cDNA fragments of zebrafish vasa, nanos2, sycp3, amh1, and insl3 were amplified by reverse transcription-PCR using gene-specific primers (Table 2) and then cloned into the pCS2P+ vector. After linearization of plasmid DNA by restriction enzymes, antisense strands were transcribed in vitro using the DIG RNA labeling kit (Roche). Paraformaldehyde (PFA) and methanol fixed testes were cut into ∼1 mm blocks and treated with proteinase K (20 μg/mL; Roche) for 20 min at room temperature. Hybridization was performed overnight at 65°C. After hot washes, testis blocks were immunostained with a mouse anti-DIG antibody conjugated with alkaline phosphatase (AP; Roche) diluted 1:2000. The 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium (BCIP/NBT) AP substrate kit from Vector Laboratories was used for coloration following the manufacturer's guideline. After coloration, the testis blocks were postfixed in 4% PFA, embedded in Tissue-Tek OCT compound, and sectioned at 6 μm thickness for imaging. After mounting on glass microscope slides, images were taken by the Olympus BX51 microscope equipped with a color CCD camera (DP72; Olympus).
List of Primers Used for Generating RNA Probes
BamHl and Xbal sites (underlined) were added for cloning purposes.
Statistical analysis
Statistical analysis was performed using the GraphPad program (https://graphpad.com/). Unpaired t-tests were used to determine the significance of the difference in the means for two groups. p Values of ≤0.05 were considered statistically significant.
Results
Testicular tumor formation in zebrafish ns1402 mutants
ns1402 mutants were spontaneously isolated from the wild-type AB zebrafish colony in our facility. Gene(s) that might be responsible for the testicular tumor formation in ns1402 mutants have not yet been identified. ns1402 mutants were maintained by single-pair mating either between prospective ns1402 mutant carrier female and male zebrafish, or between prospective ns1402 mutant carrier female and wild-type AB male zebrafish. At the time of mating, testicular tumors had not developed in ns1402 male zebrafish. If their progenies developed testicular tumors by 15 months of age, we considered the parent fish to be carriers of the ns1402 mutation. These fish were assumed to carry either heterozygous or homozygous ns1402 mutation, and we call them ns1402 mutants hereafter.
The average incidence rate of testicular tumors in ns1402 mutant males was 59% at 1.4–1.9 years of age (Fig. 1A). This was significantly higher than that in wild-type zebrafish males, which was 2% in both a previously published study 14 and in AB wild-type zebrafish in our facility (Fig. 1A). ns1402 mutant males were fertile at young age and did not show significant defects in gross morphology or histology of the testes (data not shown). At 9 months of age, visible tumors started to develop in ∼20% of ns1402 mutant males and the incidence of the tumors increased with age (Fig. 1B). We confirmed that the high incidence of testicular tumors was inherited for at least three generations (Fig. 1C).
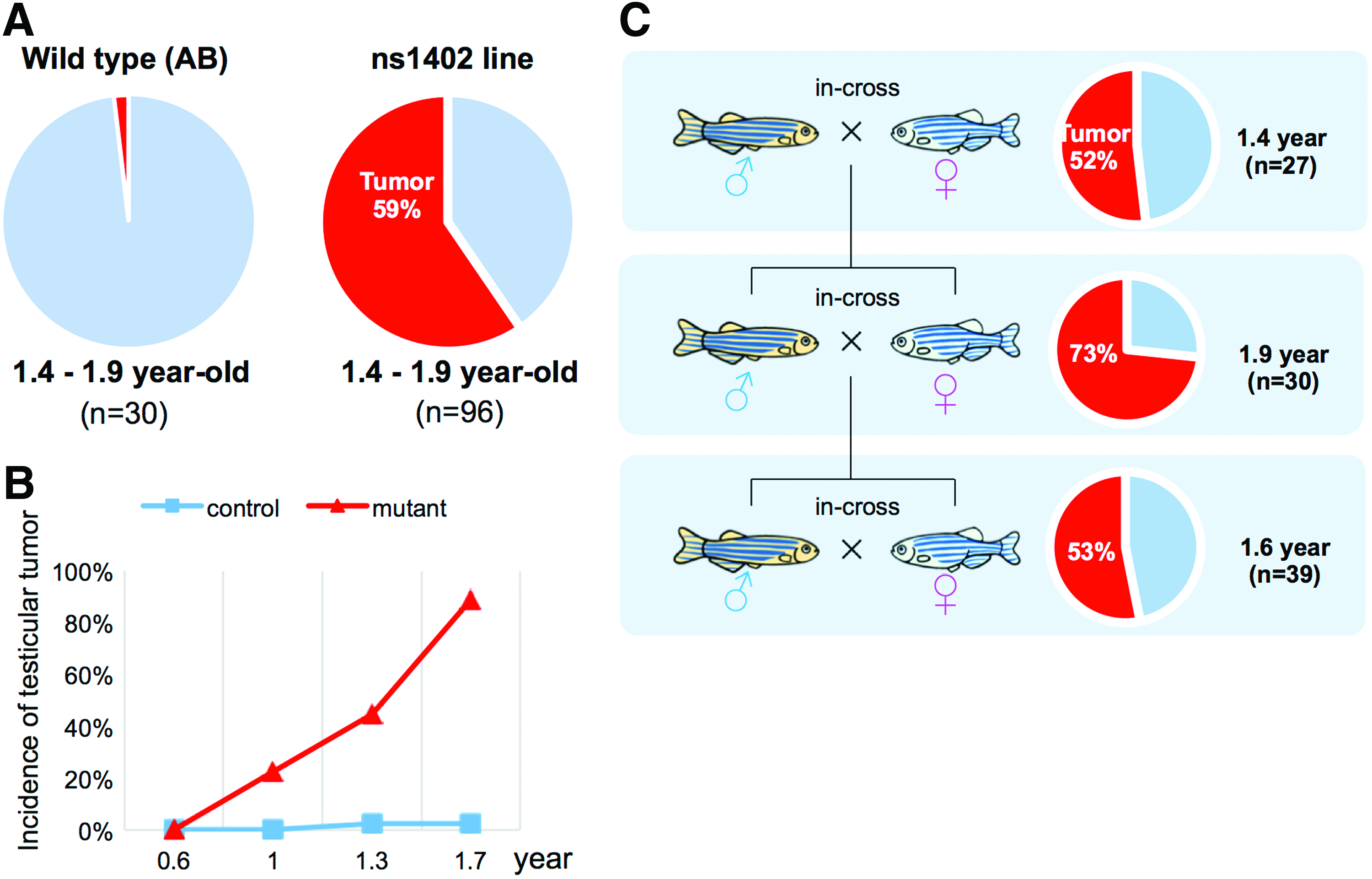
Identification of ns1402 mutants that developed testicular tumors.
We categorized tumors in ns1402 mutants into three stages based on gross morphology (Fig. 2A–D) and histology (Fig. 2E–P) of the testes. In the normal wild-type testes, individual seminiferous tubules were surrounded by the basement membrane (Fig. 2M). Sertoli cells formed spermatogonial cysts containing clusters of spermatogonial germ cells at the same differentiation stage. 15 Mature sperms were localized at the center of the seminiferous tubules (Fig. 2E, M). In ns1402 mutants, the early-stage tumors had seminiferous tubules that were smaller and the basement membrane was disorganized (Fig. 2N). Intense nuclear staining derived from mature sperm was also lost (Fig. 2F, N). The mid-stage tumors were highly vascularized (Fig. 2C), lacked distinctive seminiferous tubule structures (Fig. 2O), and contained only cells with large round nuclei and triangular cells (Fig. 2K). The late-stage tumors were translucent (Fig. 2D) and contained a large number of empty cysts with no cells (Fig. 2H). The number of tumors that were categorized into the mid-stage or late-stage tumors increased progressively with age (Fig. 2Q). This suggests that these tumors are progressive forms of the same tumors.

Formation of testicular tumors in ns1402 mutants.
ns1402 mutants developed TGCTs mixed with Sertoli cells
We next determined cell types overpopulating ns1402 mutant testes. In zebrafish testes, spermatogenesis proceeds within the spermatogonial cysts formed by Sertoli cells.15–18 Single undifferentiated spermatogonia type A (Aund) are the spermatogonial germ stem cells. They are associated with one or two Sertoli cells at the basement membrane of the seminiferous tubules. Spermatogonia Aund proliferate slowly and develop into differentiated spermatogonia A (Adif) and then spermatogonia B. Spermatogonia type B proliferate rapidly and differentiate into spermatocytes. After the Sertoli cell barrier formed by tight junctions is established, spermatocytes undergo meiosis and differentiate into spermatids and then spermatozoa. 17 Spermatozoa are released into the adluminal compartment of the seminiferous tubules.15,17,18 Spermatogonial cysts play a central role in providing spermatogonial germ cells with mechanical support and factors essential for spermatogenesis.15,17,18
While the seminiferous tubules of the normal testes contained clusters of spermatogonial germ cells at all the differential stages (Fig. 2E, I), the mid- and late-stage tumors in ns1402 mutants contained only two cell types: large round cells and small triangular shaped cells (Fig. 2K). We thought that the large round cells were likely to be spermatogonia. 15 We confirmed the increase of spermatogonia by in situ hybridization of spermatogonial markers (Fig. 3A–K), such as vasa19,20 and nonos2.21–23 We further confirmed the increased population of spermatogonia quantitatively by reverse transcription-quantitative PCR (RT-qPCR) of vasa, nanos2, and gfra1 (Fig. 3M–O).
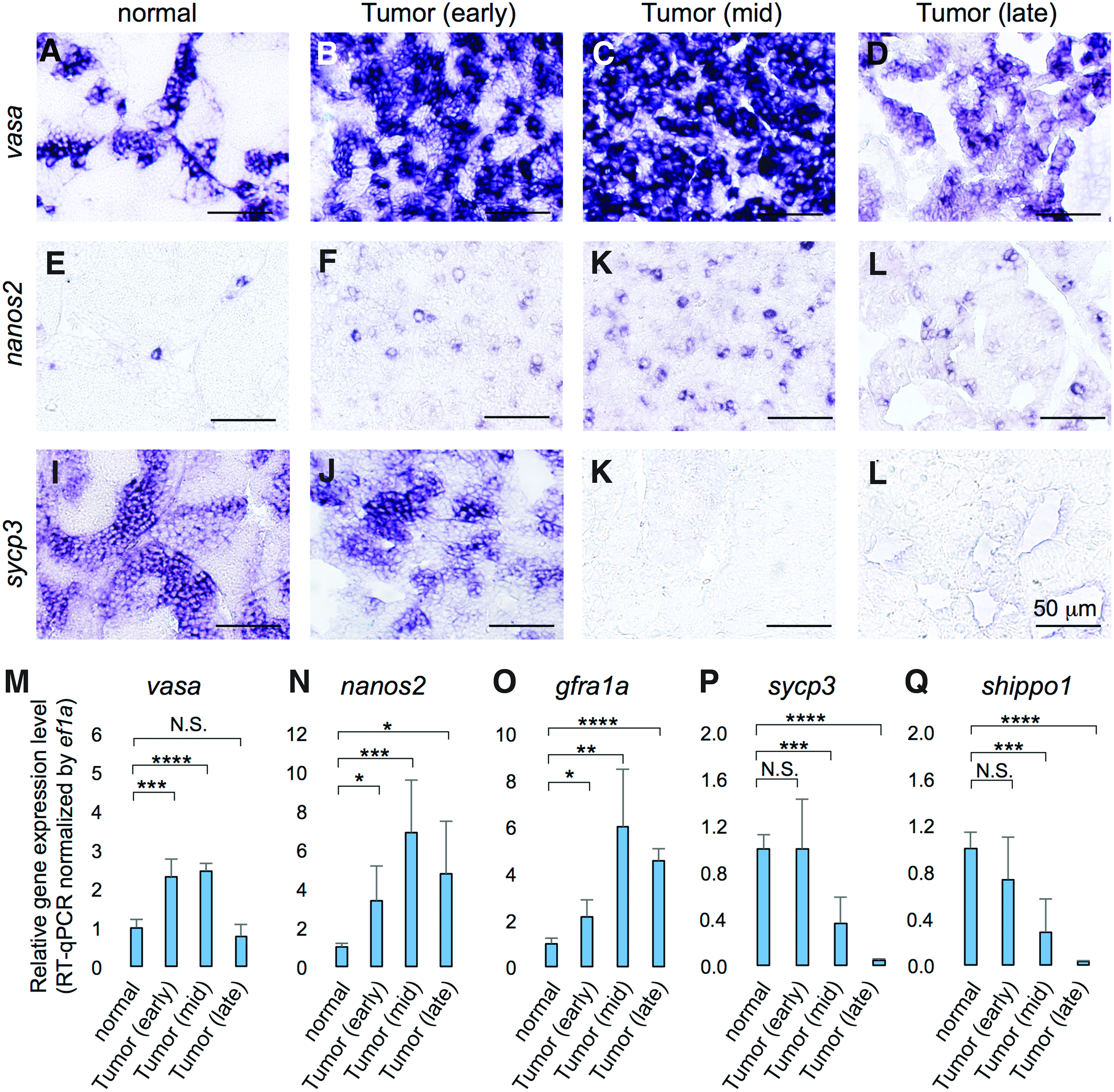
The progressive failure of spermatogenesis in ns1402 mutant testes.
To determine subtypes of spermatogonia, we used morphology of the nuclei since definitive molecular markers to distinguish spermatogonia type Aund, Adif, and B have not been established in zebrafish. The analysis revealed that spermatogonia type Aund and Adif progressively became more dominant in ns1402 mutant tumors (Fig. 4). The increase of less differentiated spermatogonia was accompanied by progressive loss of spermatocyte and spermatid marker gene expression (Fig. 3I–L, P, and Q), such as sycp317,18,24,25 and odf3b/shippo1.17,26 While mature sperms were already absent at the early stage of tumor formation (Fig. 2F, J, and N), the expression of spermatocyte or spermatid marker genes was not altered at that stage (Fig. 3J, P, and Q). This suggests that defects in spermatogenesis in ns1402 testes were initiated by failure of mature sperm production, followed by failure of spermatocyte and then spermatogonia type B differentiation.
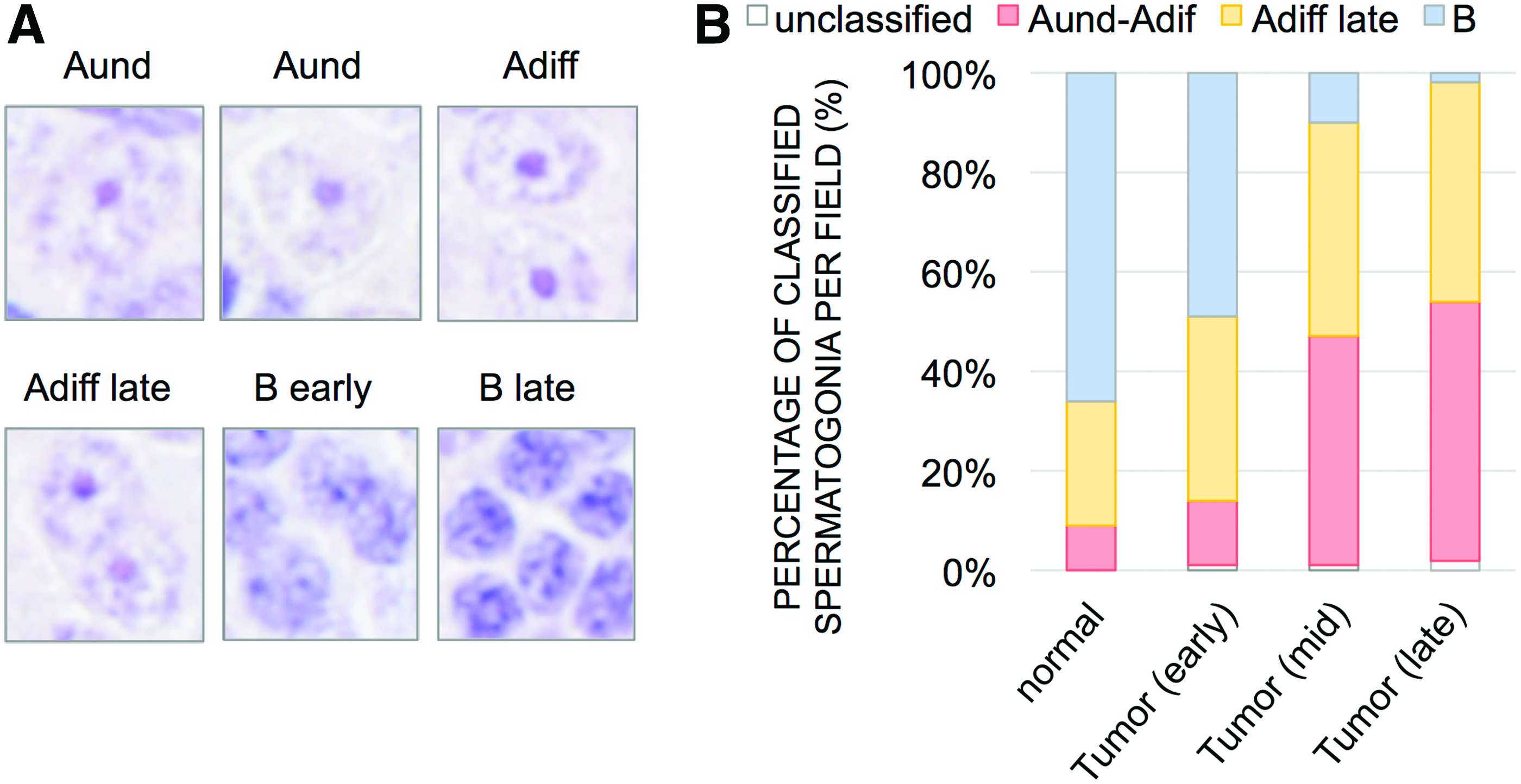
Progressive increase of undifferentiated spermatogonia in ns1402 mutant testes.
The other cells observed in the mid- and late-stage tumors showed triangular shapes (Fig. 2K, L), which we thought were likely to be Sertoli cells. The increase of Sertoli cell population was confirmed by in situ hybridization (Fig. 5B–D), RT-qPCR (Fig. 5E–G), and immunohistochemistry (Fig. 5J–M) of Sertoli markers, such as amh, 27 sox9a, 27 and wt1a.28–31 We further confirmed that Sox9 protein was accumulated in the triangular shaped nuclei in ns1402 mutant tumors (Fig. 5O). Taken together, these results suggest that ns1402 mutants developed TGCTs consisting of undifferentiated spermatogonia type A and Sertoli cells.
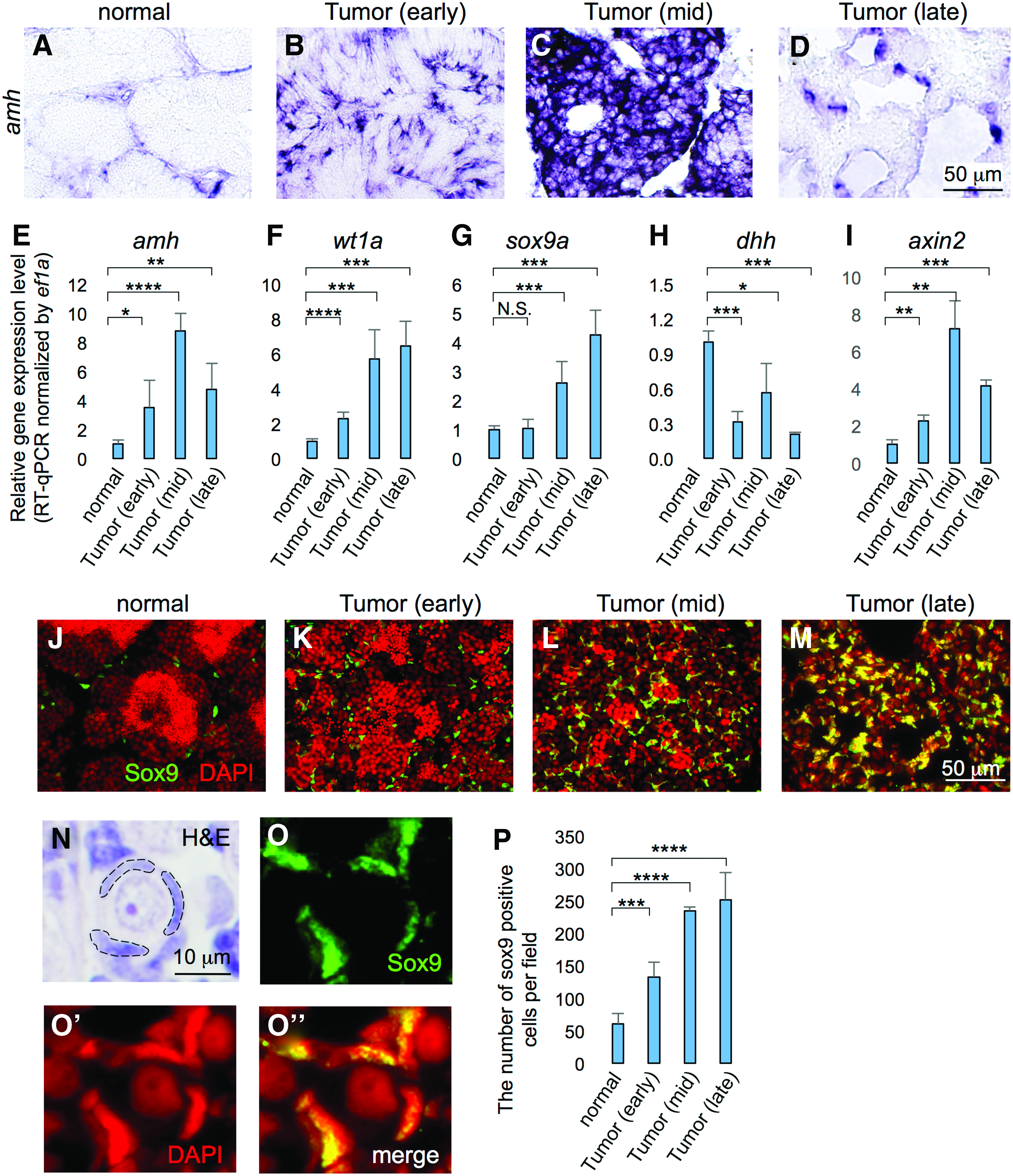
Progressive increase of the Sertoli cell population in ns1402 mutant testes.
Progressive loss of Leydig cells in ns1402 mutant testes
Next we asked the underlying causes of the impaired spermatogenesis in ns1402 mutant testes. It has been shown that testosterone derived from Leydig cells is essential for spermatogenesis. Sertoli cells are the major cells that have receptors for testosterone in the testes. 32 Testosterone activates androgen receptor (AR) and controls expression of downstream target genes. It may also activate kinases required for the progression of spermatogenesis. 33 In the absence of testosterone or AR, spermatogenesis fails to proceed beyond meiosis.33–35 Since the number of mature sperm was already significantly reduced in early-stage TGCTs in ns1402 mutants (Fig. 2F, J, and N), it was possible that the level of testosterone might be downregulated in ns1402 mutants.
Instead of measuring the level of testosterone directly, we examined the expression of cyp17a1, an essential enzyme for the synthesis of testosterone in Leydig cells.19,36–38 At 6 months of age, there was no change in cyp17a1 expression between prospective ns1402 mutants and their wild-type siblings (data not shown). This was consistent with normal fertility and histology of the testes in ns1402 mutants at that age. Interestingly, cyp17a1 expression was already downregulated in early-stage TGCTs in ns1402 mutants (Fig. 6J). The expression of other mature Leydig cell markers, such as insulin like 3 (insl3)18,39–41 and steroidogenic acute regulatory protein (star),17,42,43 was also downregulated (Fig. 6B, F, and I–K). On the other hand, LIM homeobox gene 9 (lhx9), a factor expressed in undifferentiated interstitial cells,44–47 was increased in mid- and late-stage TGCTs (Fig. 6L). Taken together, these results suggest that loss of mature Leydig cells likely contributed to failure of sperm production in ns1402 mutant testes.
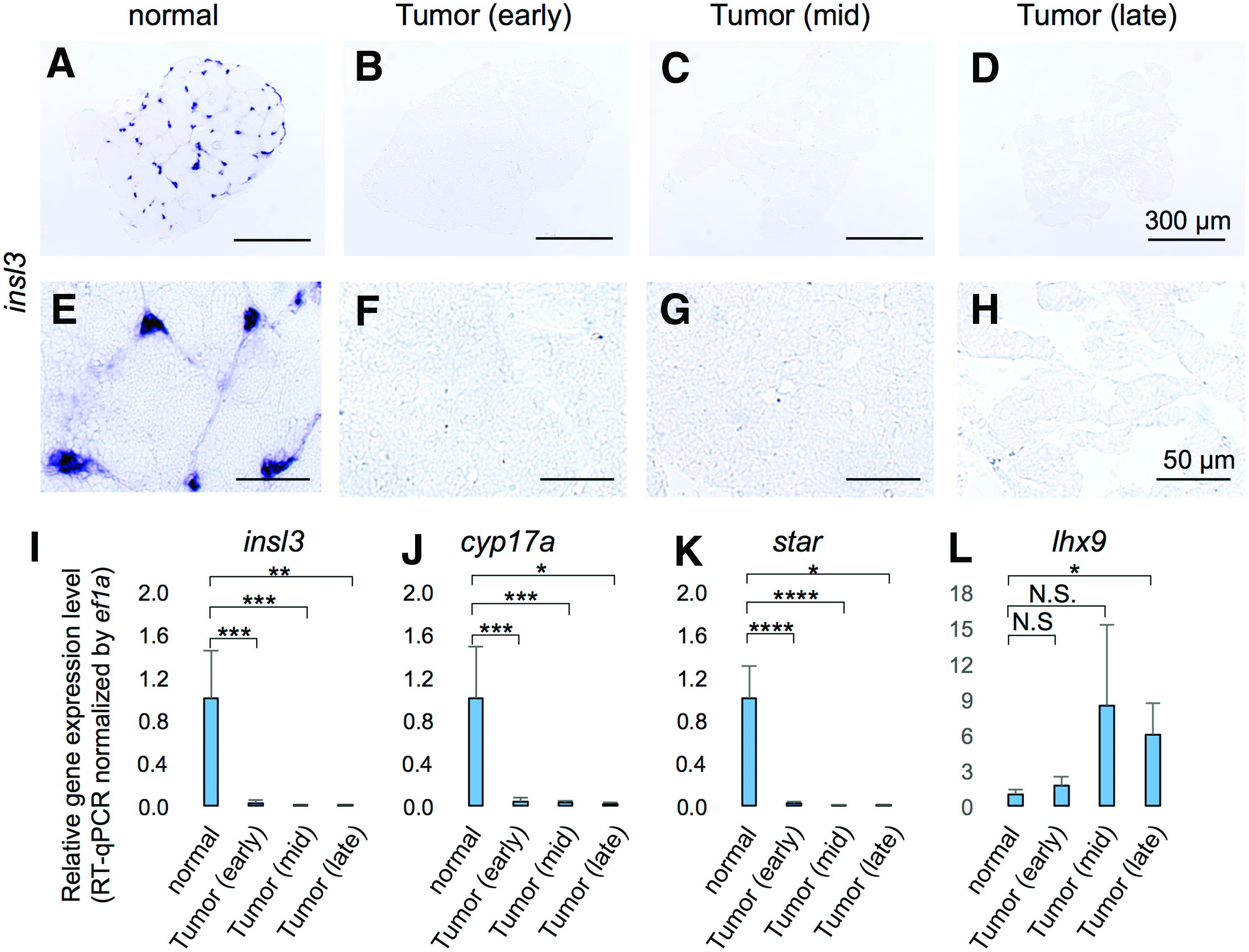
Progressive loss of mature Leydig cells in ns1402 mutant testes.
Sertoli cells became functionally less mature in ns1402 mutant testis
Leydig cells secrete growth factors and hormones, such as testosterone, which activate signal transduction pathways and control the expression of downstream target genes in Sertoli cells. This raised the possibility that the loss of Leydig cells in ns1402 mutants might modulate the activation of signal transduction pathways in Sertoli cells, resulting in failure of Sertoli cells to provide adequate environments for spermatogenesis. To test the possibility, we examined the expression of various Sertoli marker genes.
Anti-Müllerian Hormone (AMH) is a paracrine glycoprotein hormone secreted by Sertoli cells, which inhibits steroidogenesis in Leydig cells. 48 Downregulation of amh is essential for spermatogenesis. 49 RT-qPCR analysis revealed that the expression of amh increased during the early stage of TGCT formation (Fig. 5E). This increase in amh expression might be due to either increased amh expression in individual Sertoli cells or increased number of Sertoli cells expressing amh in the testes. In situ hybridization analysis showed that the number of Sertoli cells expressing amh seemed to be elevated in the early- and mid-stage TGCT in ns1402 mutants (Fig. 5B, C).
On the other hand, other Sertoli-specific genes showed different gene expression profiles. wt1a expression was increased in early-stage TGCTs and the expression was further increased in mid- and late-stage TGCTs (Fig. 5F). sox9a expression was not altered significantly in early-stage TGCTs (Fig. 5G). While sox9a expression was increased in mid- and late-stage TGCTs (Fig. 5G), the increase was not as much as the increase in amh or wt1a expression (compare Fig. 5E–G). Interestingly, the number of cells expressing Sox9 protein was already increased in early-stage TGCTs (Fig. 5K, P). It should be noted that the levels of Sox9 expression seemed to be highly variable in individual Sertoli cells (Fig. 5K–M). These results raised two possibilities, that Sox9 protein might be stabilized in Sertoli cells in ns1402 mutant TGCTs and that Sertoli cells in ns1402 mutant TGCTs might not be homogenous. Further investigation will be necessary to address these possibilities.
Desert hedgehog (Dhh) is another paracrine factor derived from Sertoli cells, which facilitates differentiation of Leydig cells during testicular development.47,50 In RT-qPCR analysis, dhh expression was already downregulated in early-stage TGCTs (Fig. 5H), suggesting that the Dhh signaling pathway was downregulated. Previous studies showed that dysregulation of Wnt signaling was involved in the pathogenesis of testicular tumors.51–56 Overactivation of Wnt signaling in Sertoli cells disrupted differentiation and promoted proliferation of Sertoli cells, resulting in testicular Sertoli tumors.52,54 Since there was an increased population of Sertoli cells in ns1402 TGCTs, it was possible that Wnt signaling might be hyperactivated. The expression of axin2, a direct downstream target gene of Wnt signaling, was indeed increased in ns1420 mutant TGCTs (Fig. 5I). Taken together, these results suggest that Sertoli cells became functionally immature and underwent deregulated proliferation in ns1402 mutant TGCTs.
sox9a and dhh were downregulated in ns1402 mutant testes before TGCT formation
ns1402 Mutants had phenotypes with loss of mature Leydig cells and an increase in immature Sertoli cell population at the early stage of TGCT formation. This raised the question of which occurred first. To address this, we analyzed gene expression profiles in the testes in which TGCTs had just begun to develop in ns1402 mutants (Fig. 7). In this analysis, normal testes were the testes that did not show visually distinguishable tumor formation on either side. A testis with a primary tumor (red arrowhead in Fig. 7A) and a non-tumor testis (white arrowhead in Fig. 7A) were isolated from one zebrafish. We confirmed that the non-tumor testis showed relatively normal histology and contained mature sperm (Fig. 7B). On the other hand, the testis with a primary tumor showed mixed histology (Fig. 7C). The seminiferous tubules in a part of the testis showed relatively normal histology and contained mature sperm, while the seminiferous tubules in the other part of the testis (an asterisk in Fig. 7C) were severely disorganized and contained no mature sperm.
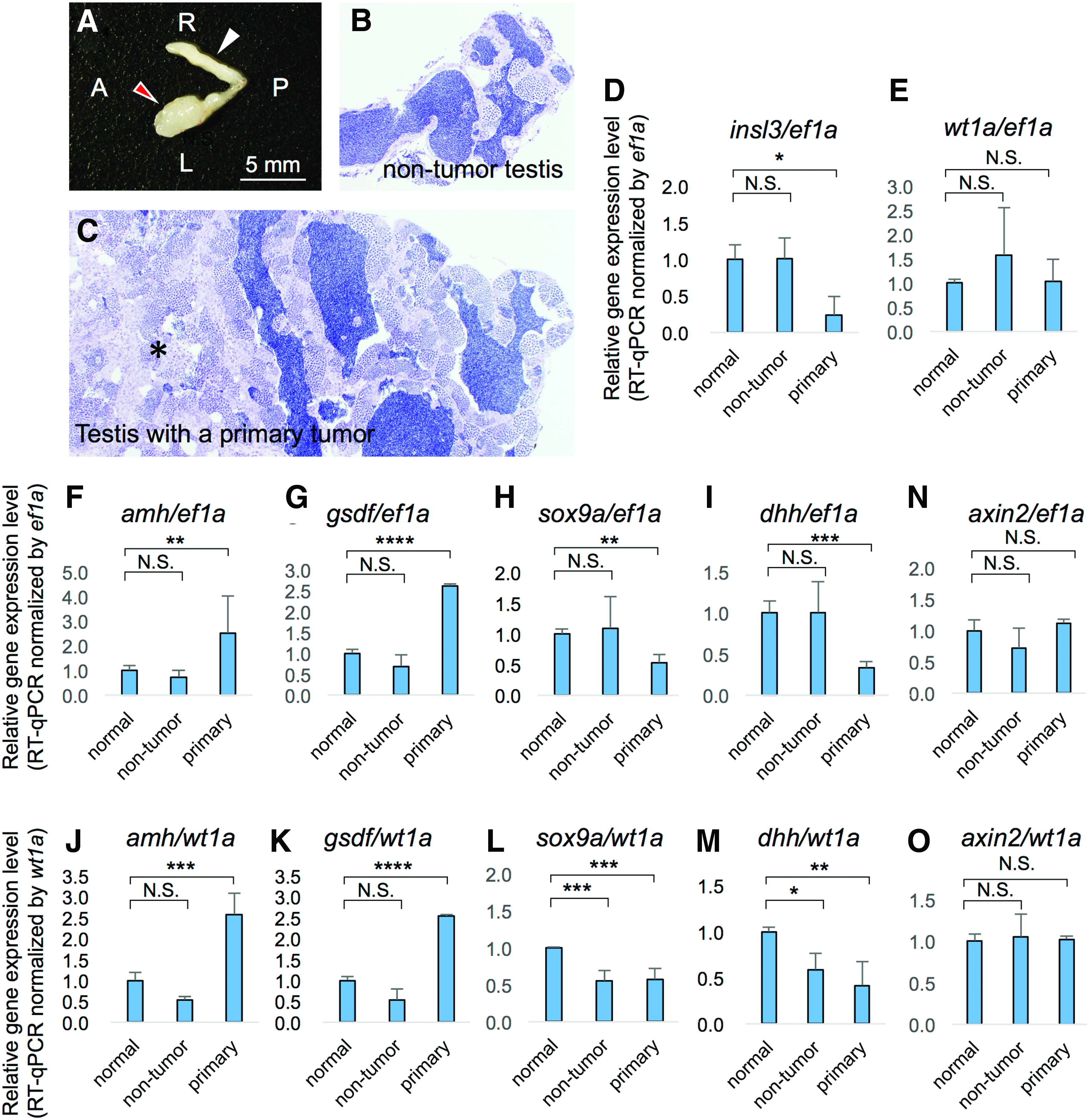
sox9a and dhh1 expression were decreased in ns1402 mutant testes before loss of Leydig cells.
mRNA was extracted from these individual testes and gene expression was quantitatively analyzed using RT-qPCR. First, we examined the expression of Leydig cell markers. The expression of insl3, a marker of mature Leydig cells, was decreased in testes with primary tumors, but not in the non-tumor testes from the same fish (Fig. 7D). This suggests that ns1402 mutant testes progressively lost mature Leydig cells during TGCT formation. On the other hand, Sertoli cell markers showed diverse gene expression patterns before and during TGCT formation. sox9a and dhh1 expression were decreased in testes with primary tumors, but the expression was not altered in the non-tumor testes (Fig. 7H, I). This was consistent with the reduced expression of sox9a and dhh in the early-stage TGCT in ns1402 mutants (Fig. 5G, H). In contrast, amh and gsdf expression were increased in testes with primary tumors, but was not in the non-tumor testes (Fig. 7F, G). wt1a expression did not differ between testes with the primary tumors and the non-tumor testes (Fig. 7E).
It should be noted that the degree of TGCT formation seemed to be variable among individual ns1402 mutant testes, especially at the initial stages of TGCT formation. The expression of Sertoli markers was variable in the non-tumor testes (wt1a in Fig. 7E, sox9a in Fig. 7H and dhh in Fig. 7I). This suggests that, while the morphology of the non-tumor testes looked relatively normal, Sertoli cell function might have been affected in these testes as well. The number of Sertoli cells had already increased at the early stage of TGCT formation (Fig. 5B, K, and P). Therefore it was important to determine whether increased gene expression observed in the RT-qPCR analysis simply reflected increased population of Sertoli cells in the testes or increased gene expression in individual Sertoli cells.
To quantify the expression of Sertoli marker genes in individual Sertoli cells, we normalized gene expression using wt1a. Previous studies showed that wt1 was expressed in fetal Sertoli cells and the expression continued at high levels throughout development in mice. 57 Nuclear immunostaining of WT1 was evident in Sertoli cells at all ages and showed no obvious maturation-dependent changes. 58 The expression of wt1 in Sertoli cells was also shown to be independent of Wnt signaling, 51 gata4 and nr5a1a.59,60 Therefore, the levels of wt1a expression seemed to be more stable than other Sertoli markers such as amh whose expression was shown to be downregulated during Sertoli cell maturation.48,49 After normalization by wt1a, we found that the expression of amh1 and gsdf indeed increased in individual Sertoli cells only in the testes with primary tumors, but not in the non-tumor testes (Fig. 7J–K). This suggests that amh1 and gsdf expression were increased after TGCTs started to develop. On the other hand, the expression of sox9a and dhh1 was decreased in both the testes with primary tumors and the non-tumor testes (Fig. 7L–M). This suggests that sox9a and dhh1 expression were decreased before the TGCT formation.
While Wnt signaling pathway was hyperactivated in the early stage of TGCTs in ns1402 mutants (Fig. 5I), there was no significant change in axin2 expression either in testes with primary tumors or the non-tumor testes (Fig. 7N, O). This suggests that Wnt signaling was hyperactivated during TGCT formation, not before TGCT formation. Taken together, these results suggest that Sertoli cells changed their property first, followed by loss of Leydig cells in ns1402 mutant testes.
Discussion
In this study, we identified ns1402 mutants, which developed TGCTs consisting of undifferentiated spermatogonial germ cells and Sertoli cells. While TGCTs mixed with Sertoli cells have been reported in humans as well as in dogs,61–65 incidence might be extremely rare. Indeed, the most recently published guideline for human cancer classification has removed mixed germ cell-stromal cell tumors from testicular tumors. 66 Nevertheless, our analysis showed that TGCTs in ns1402 mutants contained a large population of Sertoli cells. It would be interesting to determine whether the TGCTs mixed with Sertoli cells in ns1402 mutants are unique to fish. It would also be interesting to determine whether TGCTs in fish are usually mixed with Sertoli cells.
While the gene mutation(s) responsible for TGCT formation has not been identified in ns1402 mutants, no significant loss of prospective ns1402 mutant embryos or juvenile fish was observed. This suggests that homozygous ns1402 mutants might be viable and that TGCT incidence might be high in both heterozygous and homozygous ns1402 mutants. The progressive loss of fertility and TGCT formation in ns1402 mutants were associated with progressive loss of Leydig cells and increases of premature Sertoli cells. While our results suggest that Sertoli cells changed their cell properties first, followed by loss of Leydig cells, the loss of mature Leydig cells might contribute to further changes in Sertoli cell properties and then failure of spermatogenesis.
In our analysis, downregulation of sox9a and dhh was the earliest observed change in ns1402 mutant testes. Given this, the next question could be what the roles of Sox9a and Dhh in regulating spermatogenesis in adult testes are. Another question could be to ask what caused downregulation of sox9 and dhh in ns1402 mutant testes. Previous studies showed that DHH signaling is required for the differentiation of fetal Leydig cells as well as the maintenance of gene expression required for steroidogenesis in developing gonads. Failure of DHH signaling led to abnormal testicular development and infertility.47,67–71 However, function of DHH signaling in adult testes is not fully understood. Both Patched (PTCH) receptors and downstream targets of HH signaling are expressed in germ cells, and their expression dynamically changes during spermatogenesis in rodent testes.72–74 Studies also showed that DHH signaling promotes survival of germs cells and that the downregulation of DHH signaling is required for the formation of early condensing spermatids.72,73,75 dhh expression was also suppressed by follicle-stimulating hormone (FSH) and receptor tyrosine kinases in vitro.68,73 Therefore, FSH level might be increased in ns1402 mutants, which could lead to the downregulation of dhh in Sertoli cells.
Sox9, sex-determining Region Y (SRY)-9, is a member of the high-mobility group-box class of transcription factors, and controls the expression of genes required for stem cell maintenance, proliferation, and survival.76,77 Sox9 is also involved in tumor formation in various organs. For example, dysregulation of Sox9 has been reported in breast cancer 78 and pancreatic acinar cell carcinoma.79,80 In developing gonads, Sox9 function has been well characterized during sex determination and gonad development. sox9 deficiency before the key stages for sex determination led to sex reversal from male to female.81,82 sox9 expression was initiated and maintained by the combination of Sry and Sf1 transcription factors. 83
Sox9 function in adult testes is less well understood. Sox9 functions, together with Sox8, to prevent genetic reprogramming from testes to ovary. Ablation of both sox9 and sox8 led to transdifferentiation of Sertoli cells to granular cell-like cells in mice. 84 Since sox9 expression persists in Sertoli cells throughout life even after the transient Sry expression disappears in developing gonads, other factors seem to be required to maintain sox9 expression in adult testes. Recent studies showed that sox9 expression was upregulated by the fibroblast growth factor signaling pathway,85,86 prostaglandin D2,87,88 as well as by Sox9 itself. 89 The increase in Sox9 protein expression seemed more robust than the increase in sox9a transcripts, especially in early-stage TGCTs in ns1402 mutants (compare Fig. 5G, P). This raises the possibility that Sox9 protein might be stabilized in Sertoli cells in ns1402 mutants by uncharacterized mechanisms. This stabilized Sox9 protein may promote its own transcription in later stage TGCTs.
We observed cell property changes in Sertoli and Leydig cell populations in ns1402 mutants. The next question was how these changes affected spermatogenesis during TGCT progression. In early-stage TGCTs, mature sperms failed to be produced (Fig. 2F, J). We explained that this was due to loss of testosterone in the seminiferous tubules. In mid- and late-stage TGCTs, differentiation to spermatocytes and then to spermatogonia type B was also impaired. This progressive failure might be a result of progressive loss of essential growth factor signaling pathways for promoting spermatogonial differentiation. This is not surprising since Sertoli and Leydig cells both secret paracrine factors to control proliferation and differentiation of germ cells in the seminiferous tubules.
The loss of more differentiated germ cell populations might also be a result of the activation of signaling pathways that promote apoptosis before they are able to differentiate. In mid-stage TGCTs, Sox9-positive Sertoli cells were still associated with clusters of germ cells characterized by intense nuclear staining (Fig. 5L). In late-stage TGCTs, however, these clusters of germ cells disappeared and Sertoli cells formed their own cell clusters (Fig. 5M). This suggests that germ cells that failed spermatogonial differentiation might undergo cell death. The possibility of increased death of germ cells, especially in late-stage TGCTs, was supported by the formation of large number of empty cysts (Fig. 2D, H, and L), decreased expression of spermatogonial markers (Fig. 3M–O), and increased expression of apoptotic markers (data not shown). Failure of spermatogenesis, increased apoptosis of germ cells, and formation of Sertoli cell clusters have been reported in the literature for genetically modified mice. These mice included knockout mice of claudin-11, a component of tight junctions, 90 knockout mice of dazl, an RNA binding protein expressed in germ cells, 91 and Sertoli cell-specific knockout mice of connexin 43, a component of gap junctions. 92
While Sertoli cell population increased during early and mid stages of TGCT formation, amh1 and axin2 expression decreased in late-stage TGCTs in ns1402 mutants (Fig. 5D, E, and I). Since other Sertoli marker gene expression such as wt1a or sox9a did not decrease (Fig. 5F, G), it was unlikely that Sertoli cell population was decreased in these TGCTs. Rather, this suggests that additional changes might have occurred within Sertoli cells during the late stage of TGCT formation. These changes might further affect cell survival and differentiation of spermatogonial germ cells, which might be represented by the decreased expression of vasa, nanos, and gfra1a (Fig. 3D, H, and M–O). Further analysis will be necessary to determine the underlying mechanisms.
Proper development of germ cells is essential for the transmission of genetic information to the next generation. The precise control of proliferation, differentiation, and survival is essential. Disruption of these processes can lead to disorders, including infertility and tumor formation. However, lack of animal models has led to insufficient understanding of the molecular basis of TGCTs. Further characterization of the ns1402 mutants, in particular, identification of the gene(s) that are responsible for TGCT formation, has the potential to lead to identification of novel genetic modifiers that influence susceptibility to TGCT formation in humans.
Footnotes
Acknowledgments
We thank all members of M.M. laboratory and Dr. Ajay Chitnis (Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892) for valuable comments on the article.
Disclosure Statement
No competing financial interests exist.