
Abstract
The small characins represent a systematic puzzle in the Neotropical ichthyofauna as a result of independent miniaturization processes, adaptive convergence and lack of diagnostic characters for several genera. In order to diminish the taxonomic uncertainties and the evolutionary pathways in Hemigrammus, we carried out an integrative genetic analysis in the putatively widespread Hemigrammus marginatus Ellis, 1958 by combining cytogenetic and molecular data based on the mitochondrial Cytochrome C Oxidase subunit I (COI). Specimens of H. marginatus from the type locality in Itapicuru River basin and other two populations from coastal rivers in northeastern Brazil were analyzed and compared with the available data from other regions in South America. Conspicuous macro and microkaryotypic differences were detected between the samples from northeastern and southern Brazil (Upper Paraná River basin). Likewise, the DNA barcoding and species delimitation analyses recovered distinct Molecular Operational Taxonomical Units within H. marginatus. Therefore, the population from the type locality should be referred to as H. marginatus stricto sensu, representing a restricted characin taxon from coastal drainages (including the São Francisco River basin) along northeastern Brazil, while other populations of this small characin fish need to be taxonomically revised and managed as unique lineages.
Introduction
The Hemigrammus is one of the richest genera in Acestrorhamphidae, comprising nearly 60 valid species of small tetra fishes,1–3 including many colorful forms highly exploited in ornamental fish trade worldwide.4,5 These species represent a major issue in fish systematics because of their conserved morphology and successive events of miniaturization driven by adaptive convergence. As a result, this group of small fishes is characterized by losses or reduction of bone parts, lacking informative anatomical traits to the reconstruction of monophyletic lineages.6–8
As a matter of fact, the traditional taxonomic studies in this group are restricted to a few and usually overlapped morphological characters when compared with other tetras. For instance, the presence of an incomplete lateral line has been used to distinguish Hemigrammus from Moenkhausia (Eigenmann, 1907). On the other hand, comparative analyses proved that differences in the lateral line are effective diagnostic traits to differentiate the species from both genera. 9 In addition, Hemigrammus is recognized as an artificial (non-monophyletic) group of Characidae, historically referred to as incertae sedis. 10 Nevertheless, systematic revisions based on morphological and molecular data reallocated this and other related and taxonomically controversial genera (e.g., Astyanax, Hyphessobrycon, and Moenkhausia) in Stethaprioninae.
In spite of the recent efforts, the phylogenetic relationships of Hemigrammus and other small characids remain largely unresolved, including polyphyletic groups and several cases of synonymies.3,8,11 For instance, a comprehensive phylogenetic analysis carried out by Mirande (2019) 8 showed that Hemigrammus marginatus is more closely related to Moenkhausia, being placed in the so-called “Moenkhausia clade,” than to the congener Hemigrammus unilineatus. These data evidence how isolated morphological descriptions might lead to unrealistic estimates of the systematics and diversity of Characidae. On the other hand, phylogenomic analyses based on ultraconserved elements reliably divided the characins into four families (Spintherobolidae, Stevardiidae, Characidae, and Acestrorhamphidae), besides expanding the richness of several genera, such as Hemigrammus (currently placed in Acestrorhamphidae). 11
Taking into account the complexity, richness, and lack of detailed biological information in many small characins, studies based on integrative methodologies are recommended to reach a reliable delimitation of species or unique evolutionary lineages.6,8,12,13 In fact, this approach, especially by the incorporation of genetic analyses, has been successful to infer the potential boundaries among fish species.14–16 In the case of the Neotropical ichthyofauna, the association between chromosomal and molecular markers, such as the DNA barcode based on Cytochrome C Oxidase subunit I (COI) sequences has accurately recognized cryptic diversity and new species in megadiverse groups.15,17–19
Accordingly, Hemigrammus marginatus (Ellis, 1958) stands out as a perfect portrait of the puzzling taxonomy in neglected small characins from the Neotropical region. This species was formerly described based on specimens collected in the Itapicuru River basin, a small and coastal drainage flowing through the semiarid region in the state of Bahia, northeastern Brazil. Later, the distribution range of H. marginatus was expanded to other adjacent drainages in Bahia as well as to major South American basins (São Francisco, Paraná, and Paraguay), representing a relatively widespread taxon composed of disjunct populations found in a variety of Neotropical biomes from northeastern to southern Brazil. 20
On the other hand, differences in the color pattern among the populations of H. marginatus have been observed, as widely reported in small characins from distinct riverine systems and often misused as taxonomic traits in tetra fishes. 21 Even though caution is required for the diagnosis of species based on coloration, karyotype differences compatible with cryptic speciation processes have been reported between the populations of H. marginatus from Upper Paraná River 22 and Contas River 23 basins in southern and northeastern Brazil, respectively.
In addition, preliminary molecular analyses using the mitochondrial ATPase 6 and ATPase 8 genes identified two Molecular Operational Taxonomic Units (MOTUs) in Hemigrammus from the Upper Paraná River basin. Moreover, both MOTUs differed significantly from samples of H. marginatus collected in their type locality (Itapicuru River, Bahia), reinforcing the presence of undescribed species within this taxon. 24 Subsequently, Ota et al. (2015, 2018)25,26 suggested that H. marginatus would be a valid name restricted to the populations from basins in northeastern Brazil after recognizing errors in the description of paratypes formerly collected in rivers from Mato Grosso do Sul (Central Brazil) and other localities in South America.
Therefore, the goal of the present study was to provide useful insights about the taxonomic status and the systematics of H. marginatus based on comparative genetic analyses (DNA barcode and cytogenetics), including detailed information about populations from the type locality and other poorly studied and putatively species-rich rivers in northeastern Brazil. We also provide the first refined karyotypic characterization of H. marginatus, including the physical mapping of the 18S and 5S ribosomal genes that can be used as cytotaxonomic markers.
Materials and Methods
The specimens of H. marginatus were collected using gillnets (mesh size of 5 mm) in three points from two coastal basins in Bahia, northeastern Brazil (Fig. 1), as follows: (1) main channel of Contas River (−13.870806, −40.186673) and (2) Preto do Criciúma River (−13.999085, −39.927951) in Contas River basin, as well as (3) Itapicuru-Mirim River in Itapicuru River basin (type locality) (−11.1827557, −40.047325). The authorization for the collection of species in the wild was granted by the “Instituto Chico Mendes de Conservação da Biodiversidade” (ICMBio) under the permit SISBIO n.90490-1, and the experimental procedures were approved by the Committee of Ethics in Utilization of Animals (CEUA/UESB, n. 32/2013).
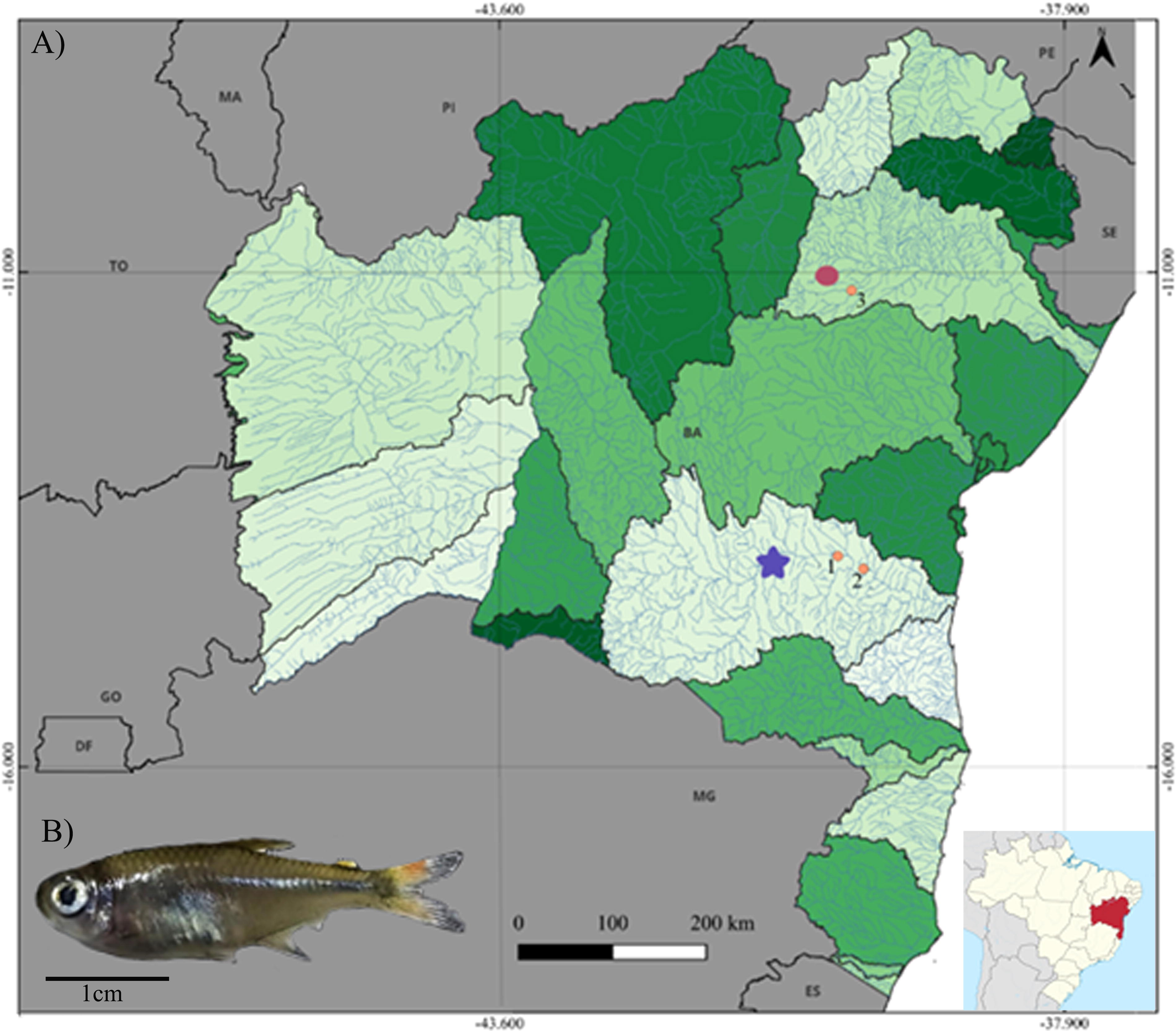
Partial map of Brazil, with focus on Bahia state showing the Contas River (blue star) and Itapicuru River (red circles) basins and the respective collection sites of Hemigrammus marginatus (specimen shown in detail), as follows: 1—main channel of Contas River; 2—Preto do Criciúma River; 3—Itapicuru-Mirim River.
Cytogenetic analysis
A total of 47 specimens of H. marginatus from both sexes were cytogenetically analyzed, being 20 of them from the Contas River (site 1), 17 from Preto do Criciúma River (site 2), and 10 samples from the type locality in the Itapicuru River basin (site 3). The animals were maintained in aerated tanks for mitotic stimulation. 27 After nearly 12 h, the specimens were euthanized in iced water, 28 and portions of the anterior kidney were used to obtain the mitotic chromosomes. 29 Voucher specimens (n. MZFS18994—MZFS18997) were deposited in the ichthyological collection from the Museum of Zoology at Universidade Estadual de Feira de Santana (UEFS).
The chromosomal morphology was determined based on arm ratio 30 using the software Easy Idio 1.0 31 and classified into metacentric (m), submetacentric (sm), subtelocentric (st), and acrocentric (a). The analyses of chromosomal microstructure were based on traditional cytogenetic methods, including conventional Giemsa staining, C-banding to the visualization of heterochromatin segments, 32 and silver nitrate staining 33 to detect the active nucleolar organizing regions (Ag-NORs). In addition, the sequential staining with the base-specific fluorochromes chromomycin A3 (CMA3), distamycin (DA), and 4’,6-diamidino-2-phenylindole (DAPI) was carried out to identify GC- and AT-rich regions, respectively. 34
The simultaneous mapping of 5S and 18S ribosomal cistrons on chromosomes of the three populations of H. marginatus analyzed in the present study was accomplished by double fluorescence in situ hybridization (double-FISH). The 18S probe was obtained according to Hatanaka and Galetti Jr. (2004), 35 and labeled by nick translation using biotin-16-dUTP (Biotin-Nick Translation Mix; Roche Applied Science), while the 5S rDNA probe was labeled via PCR with digoxigenin-11-dUTP (DIG-11-dUTP [Jena Bioscience]), using the primers 5SA and 5SB, 36 and the genomic DNA of Parodon nasus. The FISH experiments were performed under high stringency conditions (77%), and the 18S and 5S rDNA hybridization signals were detected with Streptavidin Alexa Flour 488 (Molecular Probes) and antidigoxigenin rhodamine–Fab fragments (Roche Applied Science), respectively, while the chromosomes were counterstained with DAPI (0.2 mg/mL) in Vectashield mounting medium (Vector).
The best metaphase spreads obtained in each cytogenetic technique were photographed using an epifluorescence microscope Olympus BX-51 equipped with a high-resolution digital. The fluorescent-stained chromosomal images were digitalized and overlapped using the software Image-Pro Plus® v. 6.2 (Media Cybernetics). For karyotyping, the chromosomal pairs were arranged according to each morphological class and in decreasing size order in the software Adobe Photoshop CS3 Extended v. 10.0.
Molecular analysis
The total DNA was isolated using the Wizard Genomic DNA Purification kit (Promega) according to the manufacturer’s instructions from 13 samples of H. marginatus, distributed as follows: n = 4 for site 1, n = 5 for site 2, and n = 4 for site 3. Afterward, the COI fragments were amplified via PCR using the LCO1490-F (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198-R (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) primers designed by Folmer et al. (1994). 37 Each reaction encompassed 1.5 µL of buffer (10×), 1 µL of MgCl2 (50×), 0.6 µL of each primer (10 ng/µL), 2.5 µL of dNTPs (10 mM), 0.3 µL of Taq DNA polymerase (5v/µL), 1 µL of DNA template (∼40 ng/µL) and ultrapure water to a final volume of 15 µL.
The PCR conditions were adjusted as follows: an initial denaturation step at 95°C for 4 min, followed by five cycles at 95°C for 40 s, 50°C (annealing) for 30 s and extension at 72°C for 1 min and 30 s, 35 cycles of 95°C for 40 s, 56°C for 40 s, and 72°C for 1 min and 30 s; plus a final extension step at 72°C for 10 min. The PCR products were purified in 20% polyethylene glycol (PEG), 38 washed twice in 80% ethanol, and rehydrated in the Wizard® Genomic DNA Purification solution (Promega). The sequencing reaction was performed using the Big Dye Terminator v 3.1 Cycle Sequencing kit (Applied Biosystems/Life Technologies) to a final volume of 10 µL, using 2.0 µL of 5× buffer, 0.5 µL of Big Dye, 1 µL of each primer (10 M), 1 µL of amplicon, and 5.5 µL of ultrapure water.
Afterward, the COI fragments were read on the ABI 3500 XL automatic sequencer (Applied Biosystems/Life Technologies). The high-quality COI sequences were analyzed using the “Identification Engine” tool available in the Barcode of Life Data System (BOLD) to confirm their identification, assuming only values of genetic similarity above 98%. All COI sequences obtained in this study were deposited in the BOLD platform (https://www.boldsystems.org/) under the project “Hemigrammus marginatus—UESB” (accession numbers UESB001-24 to UESB013-24) and validated as DNA barcodes (barcode index number [BIN] AAC7039).
In order to provide an accurate calibration for the comparative analysis of the genetic diversity in Hemigrammus, 78 COI sequences from closely related species available in BOLD were added based on BIN searches. In addition, nine COI sequences were also obtained from GenBank, thus totaling 87 sequences of COI from 20 genetically related taxa, identified in the public databases as: Actinopterygii, Characiformes, Astyanax bimaculatus, Curimatopsis, Hasemania sp., H. nana; Hemigrammus sp., H. boesemani, H. erythrozonus, Hemigrammus cf. gracilis, Holopristis guyanensis (formerly described as Hemigrammus guyanensis), H. hyanuary, H. levis, H. marginatus, Holopristis ocellifer, Holopristis pulcher (formerly referred to as Hemigrammus ocellifer and H. pulcher), H. rodwayi, Bario skolioplatus (formerly Hemigrammus skolioplatus), H. stictus, and Parapristella aubynei. Finally, three COI sequences from Moenkhausia bonita were included as outgroup (Table 1).
List of Species Included in the Public BINs of Hemigrammus and Outgroups Available on BOLD and GenBank Platforms (in Green) with Their Respective Accession Numbers, BINs, and Place of Origin When Informed
BIN, barcode index number; BOLD, Barcode of Life Data System.
It should be pointed out that some of the ingroup taxa were not identified as Hemigrammus but were maintained in the present database because they belonged to the BINs available in the BOLD platform for species of this genus. Moreover, the COI sequences referring to suprageneric taxonomic levels were included in the present study because they showed similarity values above 99% with Hemigrammus representatives, according to the BLAST search tool from public databases.
The COI sequences from the present dataset were edited and aligned in the software BioEdit v.7.2, using the ClustalW tool. 39 The final sequences were translated to verify the putative presence of stop codons using the software MEGA v.11. 40 A Neighbor-Joining (NJ) tree was built in the same software (Supplementary Fig. S1), based on the Kimura-2-parameter model 41 and 1000 bootstrap pseudoreplicates, 42 as preconized in studies of DNA barcoding. 43 In addition, the intra- and interspecific genetic distances were also calculated (Supplementary Table S1) to verify the presence of barcode gaps. 44
To test the reliability of the clusters, other methods of phylogenetic reconstruction based on the COI data were also carried out. First, a tree based on the Maximum Likelihood (ML) approach was generated (Supplementary Fig. S2) on the IQ-TREE online platform (http://iqtree.cibiv.univie.ac.at/) 45 using the NNI search algorithm and ultrafast bootstrap based on 1000 pseudoreplicates 46 and assuming the Jukes-Cantor (JC) substitution model, as automatically detected by ModelFinder.
In addition, a tree based on Bayesian Inference (BI) (Supplementary Fig. S3) was built using the software Mr. Bayes 3.2.6 47 available on the CIPRES Science Gateway 3.3 (http://www.phylo.org/index.php). For the BI, two independent series with four Markov chains and 10,000,000 generations each were used, assuming a burn-in of 10% and the HKT85 + G nucleotide evolution model, as indicated in the software Kakusan 4.4-0. 48 The performance of BI analysis was verified using Tracer v. 1.6 49 and the final tree was viewed and edited in FigTree v.1.4.2. 50 The summary tree was built using the software©Adobe Photoshop CC v.14.0
To confirm the potential evolutionary units within H. marginatus and determine the interspecific diversity threshold in this group, we selected only the barcodes and BINs obtained for Hemigrammus (present study and public BOLD and GenBank databases) to be analyzed based on distinct methods of molecular species identification. The tested algorithms included in this approach were: (1) Assemble Species by Automatic Partitioning (ASAP), (2) Single-Rate Poisson Tree Processes (PTP), (3) multi-rate PTP, (4) Bayesian PTP or bPTP, (4) Phylogenetic Map or PhyloMap, (5) single Generalized Mixed Yule Coalescent (GMYC), and (6) multiple GMYC.
In the case of ASAP, 51 we used a FASTA alignment file as input assuming the Kimura80 ts/tv 2.0 evolutionary model. On the other hand, an ML tree obtained on the online IQ-TREE platform (http://iqtree.cibiv.univie.ac.at/) was selected as input for the PTP 52 and mPTP algorithms available online at https://mptp.h-its.org/#/tree as well as for the bPTP and PhyloMap tests available at https://species.h-its.org/52.
In turn, the analyses using the GMYC or mGMYC 53 algorithms (https://species.h-its.org/gmyc/) were based on an ultrametric phylogenetic tree as input obtained in the software BEAST 1.8.2 54 according to the following parameters: GTR + G substitution model, strict molecular clock, arbitrary mutation rate at 1.0 substitution/site/Ma), Yule Process prior and 10 million generations sampled at every 1000 generations. In this approach, the samples are assigned into species based on the changes in branching rate from the input tree.
Results
Cytogenetic analysis
All specimens of H. marginatus analyzed shared a diploid number (2n) of 50 and a standard karyotype composed of 12m, 36sm, and 2a chromosomes, with a fundamental number (FN) of 98. In addition, all specimens presented large metacentric chromosomes in pair 1 when compared with the other pairs (Fig. 3A, B, and C). After the C-banding procedure, a conspicuous heterochromatin segment was detected occupying the entire short arms of pair 2 (m) along with centromeric C-bands widespread in most bi-armed chromosomes. Pericentromeric heterochromatin blocks were also observed in pairs 8, 16, 20, 21, and 22 (Fig. 2D, E, and F). No evidence of heteromorphic sex chromosomes was observed in the presently studied populations.

Karyotypes of Hemigrammus marginatus from Itapicuru-Mirim

Karyotypes of Hemigrammus marginatus from Itapicuru-Mirim
The major active Ag-NORs were detected at the terminal position on short arms of the second m pair (Fig. 2G, H, and I) and interspersed with GC-rich sites (CMA3+/DAPI−) (Fig. 2G, H, and I, insert). On the other hand, additional Ag-NORs were occasionally identified in the population from the main channel of the Contas River.
The physical mapping of 5S and 18S rRNA ribosomal genes in the three populations of H. marginatus from coastal rivers of Bahia (Fig. 3) revealed a conserved distribution of 5S rDNA cistrons restricted to the pericentromeric regions of pairs 18 (sm) and 25 (a). In turn, the 18S rDNA signals were mapped onto the terminal region of pair 2 (m). However, an additional and exclusive 18S rDNA signal was observed at the pericentromeric region of pair 16 in specimens of H. marginatus from the Contas River (Fig. 3B).
Molecular analysis
The analyzed COI fragments encompassed 658 base pairs (bp), with no evidence of stop codons, indicating that they actually referred to functional gene sequences. The topologies of the NJ, ML, and BI trees were identical and most branches presented support values above 97% in bootstrap or 0.9 of posterior probability (Fig. 4; Supplementary Figures S1, S2, and S3). Considering the high support values in at least two of the analyses and the congruence among most of the species delimitation methods (PTP, PhyloMap, ASAP, mGMYC, sGMYC, mPTP, bPTP, and BINs), we recovered 19 MOTUs within the present dataset (Fig. 4).

Summary tree based on NJ, ML, and BI reconstruction for the COI dataset in Hemigrammus and related taxa, showing their respective support values (>90% in bootstrap and >0.9 of posterior probabilities) on each branch. Bars on the right refer to results based on molecular species delimitation tests (BINs, PTP, PhyloMap, ASAP, GMYC-S, GMYC-M, PTP-S, PTP-M). The asterisks indicate the sequences obtained from GenBank. ASAP, Assemble Species by Automatic Partitioning; BI, Bayesian Inference; BIN, barcode index number; COI, Cytochrome C Oxidase subunit I; GMYC, Generalized Mixed Yule Coalescent; ML, Maximum Likelihood; NJ, Neighbor-Joining; PhyloMap, Phylogenetic Map; PTP, Poisson Tree Processes.
Nevertheless, some inconsistencies were observed in the number and composition of MOTUs among the species delimitation tests. While most algorithms separated this nominal taxon into five MOTUs (8 to 12), the PTP and PhyloMap grouped MOTUs 9, 10, and 11, while the ASAP grouped MOTUs 9 and 10. The intraspecific distances ranged from 0% (MOTUs 8, 9, 11, and 17) to 1.1% (MOTU 13), with a mean value of 0.3%. On the other hand, the interspecific distances varied between 4.4% (MOTUs 9 and 10) and 28% (MOTUS 2 × 13), with a mean value of 20.7%.
In the case of H. marginatus, the main focus of this study, all delimitation algorithms grouped the specimens collected in coastal rivers of Bahia with samples of H. marginatus in the BIN-AAC7039 with the highest support values (100% of bootstrap and 1 of posterior probability). These specimens referred to the MOTU 18, which includes the populations from northeastern Brazil (states of Bahia and Rio Grande do Norte) and Minas Gerais (São Francisco River basin). On the other hand, the COI sequences of H. marginatus from the Upper Paraná River basin (BIN AAC7038) were placed apart in MOTU 19 with high support values (97%–99% of bootstrap and 0.99 of posterior probability) (Fig. 4). The values of intraspecific variation within MOTUs 18 and 19 were estimated in 0.8% and 0.2%, respectively, while the genetic distance between the MOTUs was 7.3%.
Discussion
The remarkable diversity and the wide distribution of Neotropical characins, including several poorly resolved and overlooked taxa, represent a major challenge in fish systematics and conservation.8,15,17,55 Therefore, alternative approaches, such as cytogenetic and molecular markers, become particularly useful to overcome the taxonomic uncertainties in the Neotropical ichthyofauna characterized by fast adaptive radiation and speciation rates not necessarily accompanied by striking differences in phenotypic traits.15,56–58
In former Characidae, chromosomal analyses have been highly effective in recognizing species complexes, such as Psalidodon scabiprinnis and Psalidodon fasciatus. Comparative cytogenetic studies in populations of these characins from distinct hydrographic basins in South America have reported conspicuous macro and microkaryotypic differences that suggest the presence of several cryptic lineages.59,60 In fact, the divergence in karyotypic features within a putative single taxon reinforce the presence of potential reproductive barriers favorable to speciation processes, as repeatedly demonstrated in Neotropical vertebrates.61–63
In spite of their potential efficiency in cytotaxonomy, the cytogenetic studies in several groups of tropical tetras remain largely limited and underrepresented. In the case of Hemigrammus, the karyotype data are based on a few conventional techniques restricted to H. marginatus from the Upper Paraná 22 and to a recent report (about 20 years later) in samples from Contas River basins. 23
The present chromosomal information in H. marginatus comprised a variety of cytogenetic techniques to provide a detailed characterization of this species, including populations from the type locality (Itapicuru River basin in Bahia). Accordingly, the analyzed specimens presented 2n = 50, including a large metacentric pair, and no evidence of sex chromosome systems, as similarly reported in other populations identified as H. marginatus.22,23 On the other hand, the karyotype formulae were clearly differentiated between the samples from northeastern (12m + 36sm + 2a) and southeastern (10m + 34sm + 6a) Brazil. 22 These data indicate the role of pericentric inversions in the cytogenetic diversification of Neotropical fishes and as a potential driver of speciation, particularly favored by the geographic distance (about 2000 km apart) and the independent evolutionary history of each basin.
On the other hand, the heterochromatin distribution was slightly differentiated among the samples of H. marginatus from distinct collection sites since they shared a general pattern of (peri)centromeric C-bands, as commonly reported in small characins.64,65 Yet, a conspicuous heterochromatic segment was observed along the short arms of the second metacentric pair in the populations from Contas and Itapicuru basins, but absent in the specimens from Upper Paraná basin. 22 Therefore, this trait represents a diagnostic chromosomal marker in H. marginatus from coastal rivers in northeastern Brazil, being useful for cytotaxonomic inferences.
Moreover, the heterochromatin block in pair 2 of samples from both river basins in northeastern Brazil was associated with GC-rich sites (CMA3+) and interspersed with active Ag-NORs. It should be pointed out that the specimens from Contas River also presented additional NOR sites, putatively related to clastogenic agents caused by the high levels of pollution in the main channel of Contas River. 23 In contrast, the populations of H. marginatus from the Paraná River basin were characterized by the presence of GC-rich heterochromatin segments invariably located on pair 19. 22 Again, these data reinforce the differentiated karyoevolutionary pathways between the populations from distinct Brazilian regions as a consequence of their reproductive isolation, eventually leading to allopatric speciation.
In order to refine the cytogenetic characterization, many studies in Neotropical fishes have relied on the physical mapping of specific DNA regions (e.g., ribosomal genes), thus providing important insights about the genomic organization of this group that could be applied to biogeographic and systematic inferences.15,66,67 Accordingly, this approach confirmed the conserved chromosomal structure of H. marginatus from coastal rivers in northeastern Brazil, since the number and position of 5S and 18S rDNA sites in specimens from the type locality (Itapicuru river basin) and Contas River remained the same. In addition, the presence of single NORs observed in H. marginatus from Preto do Criciúma and Itapicuru-Mirim rivers was corroborated by the mapping of 18S rDNA cistrons. 23
Instead, the presence of an additional 18S rDNA site on pair 16 in the samples of H. marginatus from the main channel in Contas River confirms the presence of multiple NORs in this population, as previously indicated by silver nitrate staining. Putatively, the differences in the number and expression of ribosomal cistrons in specimens from this locality are associated with cytological alterations induced by high levels of environmental contaminants in Contas River. 23 Alternatively, it should be emphasized that rDNA regions are usually syntenic with transposable elements in fish genomes. 68 Therefore, copies of ribosomal genes linked to transposons can move dynamically through chromosomes, promoting numerical and structural variation within and between taxa, as previously reported in other small characins, like Megalamphodus eques (formerly described as Hyphessobrycon eques Steindachner, 1882), Astyanax lacustris, and the P. fasciatus complex.15,67,69
As for the chromosomal mapping of 5S rDNA cistrons, lower rates of intraspecific variations have been observed in the genome of representatives of Characidae 70 when compared with the 18S rDNA, as also observed in the present study. Therefore, the analyses of the genome distribution of 5S ribosomal genes represent a potential source of information for the diagnosis of cryptic species.15,69,71 For instance, the closely related tetras Serrapinnus heterodon and S. piaba are differentiated by the presence of four and eight 5S rDNA loci, respectively, representing an efficient cytotaxonomic marker. 66 These data are particularly relevant considering the challenging task of identifying small characins based on morphological traits, as observed in Hemigrammus.8,9,25 In fact, failures in the identification of taxonomic units can lead to biased estimates of distribution ranges and the threat status of fish species while priority areas for the conservation of biodiversity would be neglected.56,72
In this sense, Ota et al. (2015) 25 suggested that H. marginatus is a tetra species restricted to the rivers of northeastern Brazil, including the São Francisco River basin since the paratypes from Paraguay, Sapucaí, and Guaporé river basins (Upper Paraná) would actually belong to Moenkhausia and Hyphessobrycon, both closely related genera to Hemigrammus.8,13 Actually, Ota et al. (2018) 26 recognized that the putative populations of H. marginatus recorded along the floodplains in Upper Paraná (Central to Southern Brazil) corresponded to Moenkhausia species (Moenkhausia gracilima or M. bonita). In addition, it is worth revisiting the study conducted by Portela-Castro and Júlio-Jr. (2002), 22 who hypothesized that the specimens identified as H. marginatus from the Paraná basin would be a distinctive unit, as corroborated by the comparative cytogenetic analysis of the present study.
Considering the abovementioned taxonomic issues and the evidence of differentiated karyomorphs in H. marginatus, the utilization of molecular markers represents a suitable tool to discriminate species and resolve the systematic uncertainties in enigmatic groups. 73 As a matter of fact, studies based on sequencing of the ATPase 6 and 8 mitochondrial genes revealed high levels of genetic distance among the populations of H. marginatus from the type locality (Itapicuru River basin) and Upper Paraná, suggesting this taxon represents a species complex. 24
To evaluate the presence of distinct candidate species in H. marginatus, we added a comparative DNA barcode analysis based on COI sequences from populations across their distribution range and other related taxa, regarded as a reliable method to detect cases of synonyms or cryptic speciation.74,75 This approach allowed the accurate discrimination of 19 MOTUs among the 21 morphological taxa with a remarkable barcode gap (∼69x) between MOTUS (Supplementary Table S1), being much higher than the minimum threshold recommended by Hebert et al. (2004). 43 In addition, these results were highly supported by phylogenetic inferences and by most species delimitation algorithms (Fig. 4).
In general, the analyses of the COI dataset from the present study provided strong evidence of misidentification in public databases, as observed by the presence of single and highly supported MOTUs that clustered clearly unrelated or morphologically distinct species, such as Curimatopsis × H. guyanensis (MOTU 1) and Astyanax bimaculatus × H. stictus × Parapristella aubynei (MOTU 4) (Fig. 4). On the other hand, three MOTUs (9–11) were assigned to H. guyanensis, comprising samples from different regions in French Guiana. 76 Considering that the type locality of this species refers to a tributary of the Acarouany River, these results suggest that H. guyanensis refers to a species complex.
Furthermore, examples of undescribed species or imprecise identification were observed in MOTUs 8, 13, and 15, which included specimens identified only at the level of order or class, such as Characiformes (e.g., accession numbers FWAQ173-11, FWAQ174-11) and Actinopterygii (e.g., accession numbers GBMNA15666-19, GBMNA15667-19), respectively. Unfortunately, the presence of these specimens in the database as well as the lack of a higher number of reference COI library sequences accompanied by the evaluation of taxonomic experts hamper the accurate species identification in megadiverse fish groups, like Hemigrammus.2,3
Focusing on the specimens of H. marginatus, the DNA barcode separated this nominal taxon into two unique evolutionary lineages (MOTUs 18 and 19). This result was highly supported by the phylogenetic reconstruction and all delimitation tests (Fig. 4), including the BINs (AAC7038 and AAC7039) that are determined based on the genetic distances and the analysis of simultaneous clusters after searches throughout the entire database available on BOLD(74). We also highlight that the interspecific distance between the MOTUs identified within H. marginatus was equal to 7.3%, being much greater than the 2% threshold commonly proposed to separate fish species, 77 while the maximum intraspecific variation within each MOTU (0.8%) was remarkably lower than 2%. Therefore, the samples of H. marginatus from Upper Paraná (MOTU 19) and the populations from coastal rivers in northeastern Brazil and São Francisco River basin (MOTU 18) refer to well-established taxonomic units of reciprocal monophyly (Fig. 4).
Given that the evolution of the Neotropical ichthyofauna is intertwined with the biogeographic history of river basins, the rearrangements in the hydrographic systems would act as major forces molding the dispersal and diversification of fish lineages. 78 As inferred from the cytogenetic data, the large geographic distances and the long period of isolation among the basins occupied by the specimens from MOTUs 18 and 19 would have favored the accumulation of genetic and adaptive divergences. This process could result in fast speciation rates, independently on the origin of distinctive morphological traits, as commonly reported in Neotropical fishes.58,79,80
Thus, the integrative genetic data from this study corroborates the hypothesis that the species from Upper Paraná would be a distinct species, while H. marginatus should be a restricted taxon from northeastern Brazil and the São Francisco River basin.24–26 The combination of chromosomal and molecular analyses proved to be highly informative to resolve the taxonomic uncertainties in H. marginatus, providing a reference baseline for the recognition of species in Hemigrammus.
Conclusions
Since the identification of small characins, such as H. marginatus, based only on morphological traits remains a big issue in fish systematics, we encourage that cytogenetic data integrated with DNA barcoding should be extensively performed for resolving the taxonomic uncertainties in fish groups from biodiversity hotspots such as the Neotropics.
Accordingly, the present data recovered two evolutionary taxonomic units characterized by conspicuous chromosomal differences and high genetic divergence in H. marginatus from distinct regions in South America. Therefore, we state that genetic analyses should be incorporated in further taxonomic revisions of Hemigrammus to provide accurate estimates of the diversity and distribution of this peculiar group of tetra fishes.
Footnotes
Acknowledgments
The authors are grateful to the Ethics Committee for Animal Experimentation at UESB (CEUA/UESB 32/2013) and ICMBIO for the approval of the present experiments and the collection of specimens in the wild. The authors would also like to thank the anonymous reviewers for their collaboration in the presentation of this article.
Authors’ Contributions
All authors contributed to the study plan and experimental design. Material preparation and data collection were performed by M.B.F., A.T.d.S., J.d.A.B., and P.R.A.d.M.A. Data analyses were performed by M.B.F., J.d.A.B., M.R.V., and M.A. The first version of the article was written by M.B.F. and all authors commented on this draft. The final version was revised by J.d.A.B. and P.R.A.d.M.A. All authors read and approved the final article.
Data Availability
The data sets generated and analyzed in this study are available from the corresponding author upon reasonable request.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
The work was funded by the Coordination for the Improvement of Higher Education Personnel-Brazil (CAPES) (Financing Code 001) Author M.B.F. has received research support from CAPES (88887.673009/2022-00).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.