
Abstract

Brain drug delivery plays an essential role in modern drug development for the central nervous system (CNS). In the past, the pathway from new drug discovery to drug development for the CNS in the clinic was uninterrupted (Fig. 1). This is because the traditional trial-and-error method of CNS drug discovery invariably selected drugs that had appropriate properties with respect to both structure-activity relationships, that is, drug-receptor interactions, and structure-transport relationships, i.e., membrane permeation. However, modern methods of rational drug design and discovery use receptor-based high throughput screening methods with thousands of compounds. The drugs that are selected with high throughput screening solely on the basis of structure-activity relationships at the target receptor will invariably have poor membrane permeation properties. Such drugs will undergo insignificant transport through the brain capillary endothelium, which makes up the blood-brain barrier (BBB) in vivo.
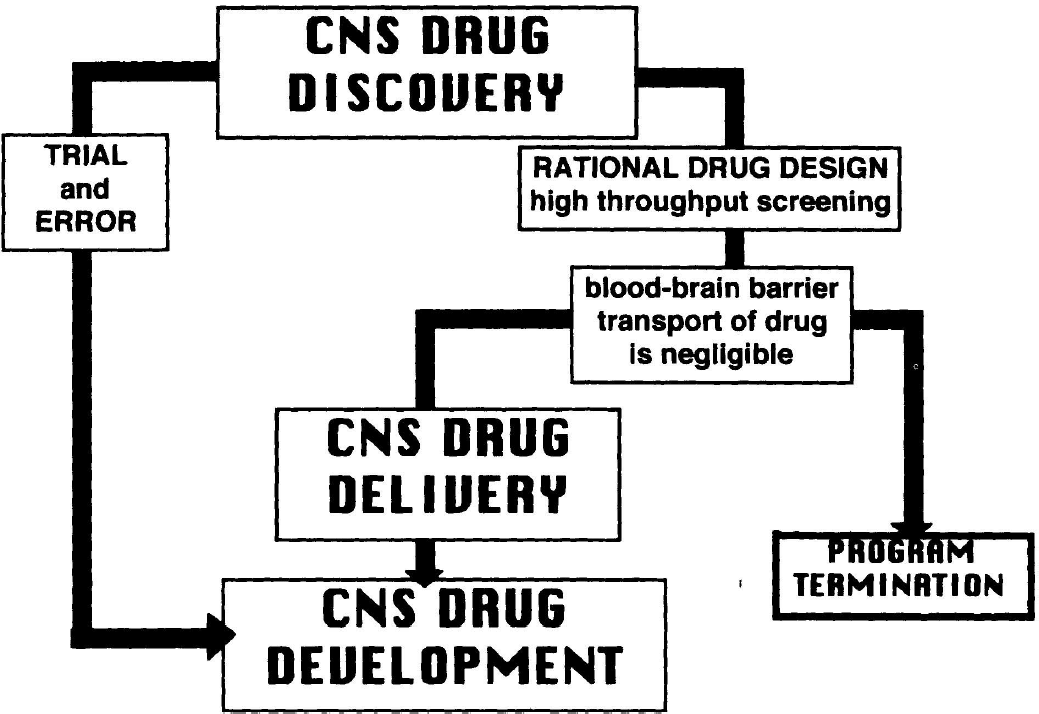
Flow chart comparing the pathways of two different methodologies for CNS drug development; the trial and error approach, and the rational drug design approach, wherein the latter is based on high throughput screening of thousands of molecules. The latter will invariably select drugs that have negligible blood-brain barrier transport properties. Therefore, in the absence of parallel progress in the development of CNS drug delivery strategies, the CNS drug discovery program invariably ends in program termination.
Modern methods of CNS drug discovery may select drugs that yield the expected pharmacologic effect after direct injection into the brain, but not after systemic administration, because BBB transport of the drug is negligible. At this stage, it is recognized that the drug has poor membrane permeation properties, and the inevitable fate of the CNS drug discovery program is termination (Fig. 1). The principal reason underlying program termination is that CNS drug discovery has evolved in the absence of the parallel maturation of an effective CNS drug delivery strategy. Indeed, given the presence of the BBB, a modern-day peculiarity is that >99% of worldwide CNS drug development is devoted to CNS drug discovery with <1% of the effort devoted to CNS drug delivery. The reason for this imbalance may lie in the fact that present-day drug-delivery science evolved from the material sciences, with a classical focus on formulation and controlled release. The material sciences give rise to brain drug delivery approaches involving intraventricular infusion, or the intracerebral implantation of genetically engineered cells or polymeric matrices. As reviewed below, these strategies do not involve drug transport through the BBB, but are invasive neurosurgical-based strategies that rely on diffusion for drug penetration into the brain. In contrast, brain drug targeting science, which aims to deliver drugs through the BBB, originates from cerebrovascular transport biology. Therefore, investigations into the fundamental mechanisms of BBB transport biology provide the platform for future CNS drug targeting strategies.
New strategies for CNS drug delivery and drug targeting are needed for at least two reasons. First, with the exception of lipid-soluble molecules, which have a molecular weight under a 400–600 d threshold, virtually all drugs that originate from either biotechnology or classical small molecule pharmacology undergo negligible transport through the BBB (Fig. 2). Second, disorders of the brain are surprisingly common, and in the United States there are >80 million individuals suffering from some form of CNS disorder, including alcohol abuse, anxiety/phobia, sleep disorders, depression/mania, drug abuse, obsessive-compulsive disorder, Alzheimer's disease, schizophrenia, stroke, epilepsy, cerebral acquired immunodeficiency syndrome, and Parkinson's disease (Pardridge, 1991).

The only class of potential neuropharmaceuticals that has significant blood-brain barrier transport are small molecules that have the dual criteria of lipid solubility, and molecular weight less than a 400 to 600 d threshold. MW, molecular weight.
The available strategies for CNS drug delivery may be broadly classified as either invasive (neurosurgical-based), pharmacologic-based, or physiologic-based (Fig. 3). The neurosurgical-based strategies include intraventricular drug infusion, intracerebral implants, and BBB disruption. The pharmacologic-based strategies include the use of lipid carriers or liposomes. The physiologic-based strategies take advantage of the normal, endogenous pathways of either carrier-mediated transport of nutrients or receptor-mediated transport of peptides.
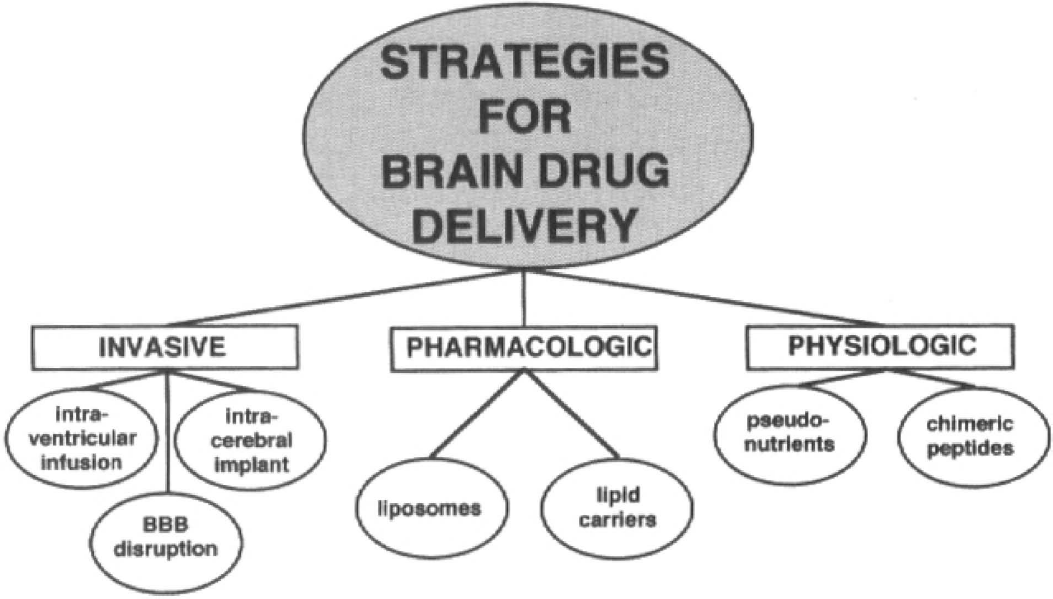
Strategies for brain drug delivery may be broadly classified as being either neurosurgical-based (invasive), pharmacologic-based, or physiologic-based.
OVERVIEW OF BLOOD-BRAIN BARRIER TRANSPORT BIOLOGY
The inability of circulating substances, even small molecules, that lack access to BBB transport systems is illustrated with histamine, which has a molecular weight of 111 d. Fig. 4A is an autoradiogram of a mouse obtained 5 minutes after intravenous injection of [14C]histamine. This small molecule readily traverses the porous capillaries in all peripheral tissues but does not enter the brain or spinal cord because of the lack of transport through the BBB. Although in the early 1900s the BBB was believed to be localized at the choroid plexus, the cellular basis of the BBB is, in fact, localized to the brain vasculature, as depicted by the scanning electron micrograph of a vascular cast of the human cerebellar cortex (Fig. 4B). The capillaries are spaced approximately 40 μm apart in the brain and it takes a small molecule approximately 1 second to diffuse a distance of 40 μm. The plasma transit time is approximately 1 second in the brain. Therefore, the angioarchitecture of the brain has evolved to allow for instantaneous solute equilibration throughout the brain interstitial fluid once the solute or drug crosses the limiting membrane, that is, the BBB. The cerebral microvasculature is comprised of four cells, as depicted in Fig. 4C. However, the permeability barrier, per se, of the BBB is comprised only of the endothelial cell, which has epithelial-like, high-resistance tight junctions that eliminate any paracellular pathways of solute flux across the endothelial barrier. Because of a paucity of pinocytosis in the cerebral capillary endothelium, there is also no significant transcellular pathway for free solute flux across the brain capillary endothelium. Therefore, a circulating molecule gains access to brain interstitial fluid only via one of two pathways: lipid-mediated transport (in the case of lipid-soluble molecules that have a molecular weight under 500 d), and carrier-mediated or receptor-mediated transport processes for nutrients and peptides, respectively. These transport systems are localized in both the luminal and abluminal membranes of the brain capillary endothelium, which are separated by approximately 300 nm of endothelial cytosol. Therefore, transport through the BBB results in movement through two membranes in series, the luminal and abluminal membranes of the endothelium. The pericyte shares a basement membrane with the endothelium and has antigen presenting properties (Pardridge, 1991). More than 99% of the brain surface of the capillaries is invested by astrocyte foot processes. However, neither these foot processes nor the basement membrane constitute a significant permeability barrier to solute diffusion in brain once the solute crosses the endothelial barrier.
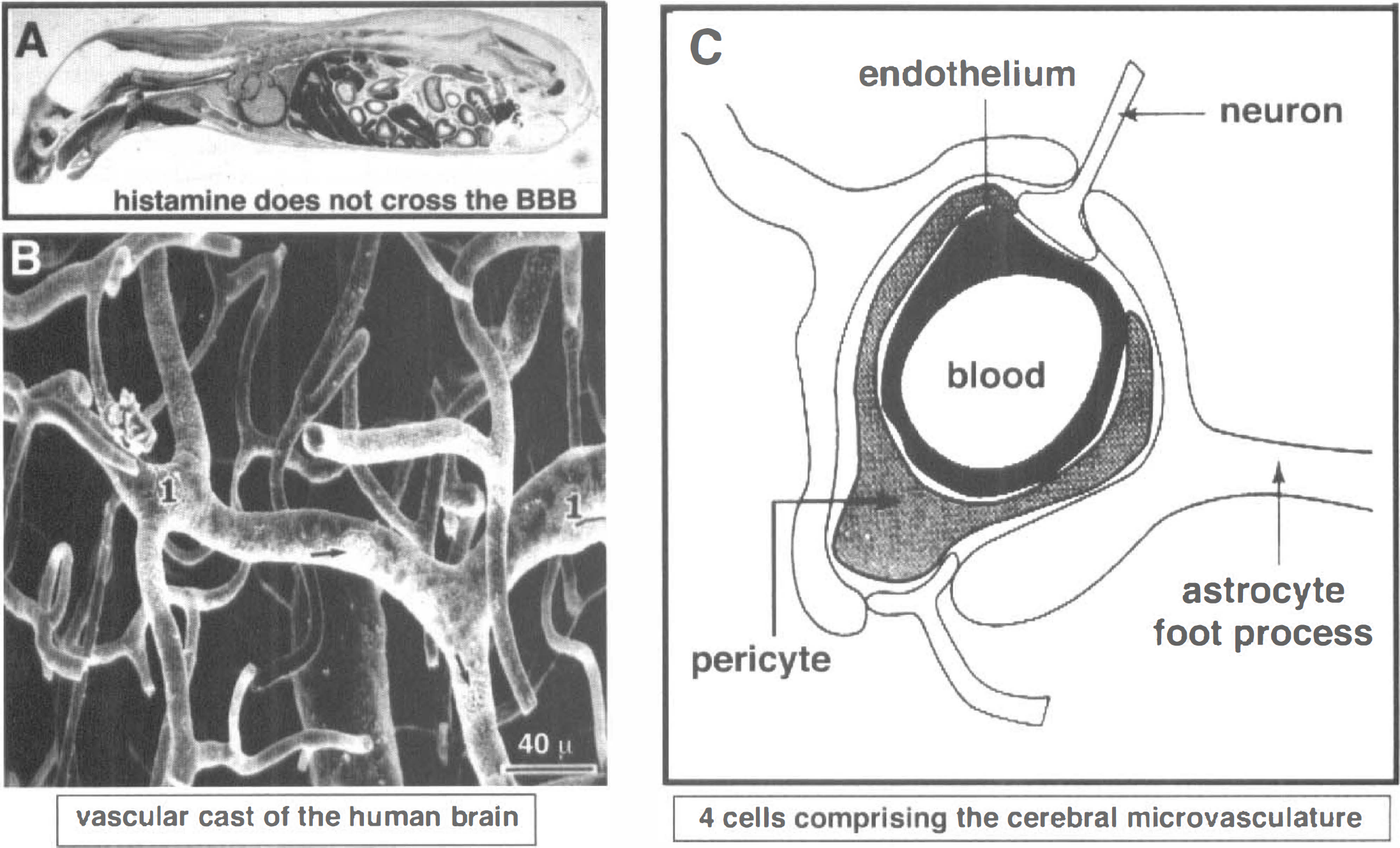
Sagittal section film autoradiogram of a mouse killed 5 minutes after intravenous injection of [14C]-histamine, a small molecule that readily crosses the porous capillary beds in peripheral tissues, but does not cross the blood-brain barrier (BBB) in brain or spinal cord (Pardridge et al., 1986).
The BBB segregates blood and brain interstitial fluid, and is a membrane separate from the blood-CSF barrier, which is formed by the choroid plexus epithelium and separates blood from CSF. The surface area of the choroid plexus or blood-CSF barrier in the human brain is 0.021 m2 (Dohrmann, 1970). Conversely, the surface area of the BBB in the human brain is approximately 20 m2 (Pardridge, 1991), a surface area that is 1,000-fold greater than the surface area of the choroid plexus.
INTRAVENTRICULAR DRUG INFUSION
Drug diffusion within the brain is limited
After the infusion of drug into the ventricular compartment, there is minimal distribution of the drug into the brain parenchyma from the ventricular or ependymal surface. The reasons for this limited drug penetration into the brain from the ventricles are twofold. First, the efficacy of diffusion decreases with the square of the distance (Jain, 1990),
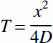
where T = time for drug diffusion through a distance (x), given a diffusion coefficient (D) in water. For example, the diffusion coefficient for a small molecule is 5 × 10−6 cm2/s (Hazel and Sidell, 1987); therefore, it takes more than 8 hours for the molecule to diffuse 1 mm. A diffusion coefficient of a neurotrophin with a molecular weight of 17,500 is on the order of 1 × 10−6 cm2/s (Pardridge, 1991), and it takes approximately 2.8 days for a molecule of this size to diffuse 1 mm. These are minimum estimates because the diffusion coefficients for molecules in brain tissue are less than the diffusion coefficients in water (Fenstermacher and Kaye, 1988). Secondly, the rate of bulk flow of CSF through the ventricles and subarachnoid space is rapid compared to the relatively slow rate of solute diffusion within the brain. There is approximately 100 mL of CSF within the human brain, and this is produced at a rate of 20 mL/h (Davson et al., 1987). Therefore, the entire CSF volume is produced and cleared every 5 hours. Cerebrospinal fluid originating in the lateral ventricle moves through the foramen to the third ventricle, through the cerebral aqueduct to the fourth ventricle, into the cisterns of the base of the brain, over the surface of the cerebellum, and finally is cleared into the peripheral blood stream via absorption at the arachnoid villi into the superior sagittal sinus. Because of this relatively rapid rate of bulk flow of CSF from the ventricles into the peripheral blood stream, the intracerebroventricular (ICV) injection of drug may be regarded as equivalent to a slow intravenous infusion of the drug. When cholecystokinin is administered ICV into the rat at a dose of 5 μg, the plasma levels of cholecystokinin reaches 2 nmol/L within 30 minutes, and the neuropeptide inhibits feeding via a peripheral mechanism (Crawley et al., 1991).
The fact that intraventricular drug infusion readily distributes drug to blood, but not to brain, has been shown repeatedly. The intraventricular infusion of [beta]-interferon in primates results in the presence of the cytokine in blood, but not in brain (Billiau et al., 1981). Similar results are found after the intraventricular injection of α-interferon in rats (Grieg et al., 1988), or 6-mercaptopurine, a small molecule, in primates (Covell et al., 1985). When a small molecule, atenolol, is administered in the left ventricle of rats, no drug is found in the dialysate of a fiber placed into the adjacent cerebral cortex (deLange et al., 1994). After the ICV infusion of acetaminophen, the drug is distributed to the peripheral circulation, is transported across the BBB, and the drug then diffuses into the dialysate of a fiber contained in the adjacent cerebral cortex (deLange et al., 1994). These results confirm earlier observations by Aird (1984) that the dose of barbiturate that induces pharmacologic effects in animals after intraventricular or intravenous injection is identical. That is, drug infused into the ventricular compartment distributes to the peripheral blood-stream, and reenters the brain via transport through the BBB.
The infusion of drug into the lateral ventricle results in distribution of drug only to the ipsilateral ependymal surface. For example, the intraventricular administration of insulin results in the detection of immunoreactive insulin only at the ependymal surface (Baskin et al., 1983). The ICV injection of a neurotrophin, brain-derived neurotrophic factor, results in neurotrophin distribution only to the ipsilateral brain surface (Yan et al., 1994), as shown in Fig. 5. That is, while intraventricular drug infusion may result in minimal penetration of drug into brain parenchyma, the ependymal surface of the CNS is exposed to very large concentrations of the drug. The intraventricular infusion of basic fibroblast growth factor results in astrogliosis in the periventricular area with increased immunoreactivity for glial fibrillary acidic protein (Yamada et al., 1991).
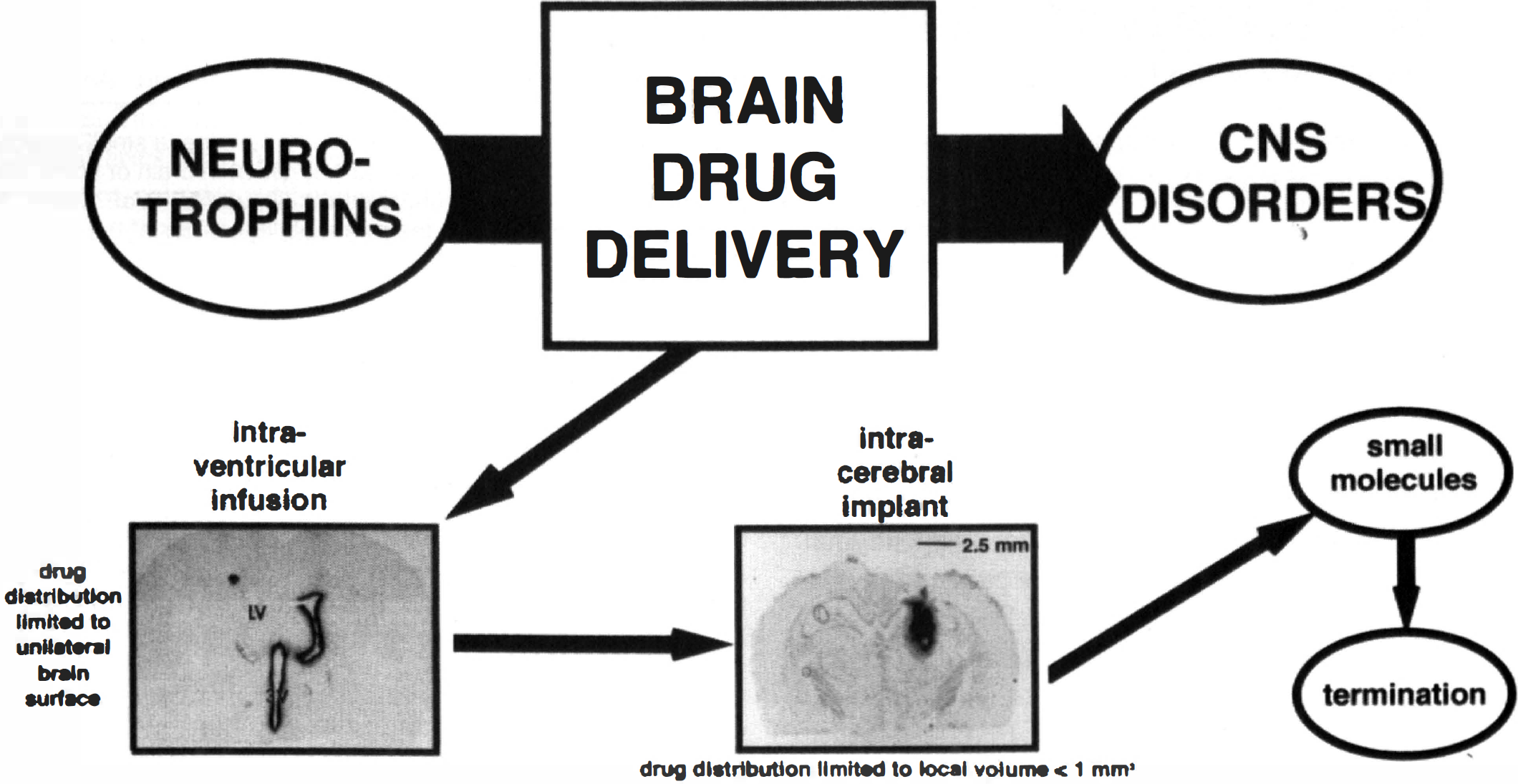
In neurotrophin drug development, the limiting factor is brain drug delivery. The strategies presently in practice for brain drug delivery in humans are outlined. The top emphasizes that the products of neurotrophin CNS drug discovery programs cannot undergo clinical development for the treatment of CNS disorders in patients, unless the brain drug delivery bottleneck is navigated. The first attempt at brain drug delivery that is generally currently tried is intraventricular drug infusion. The figure shows an autoradiogram of a rat brain taken 20 hours after the injection of [125I] brain-derived neurotrophic factor into the lateral ventricle (LV) of a rat (Yan et al., 1994). The neurotrophin is distributed only to the ipsilateral ependymal surface of the brain, which is consistent with the unidirectional flow of CSF within the brain. There is minimal penetration of the drug to the ipsilateral brain parenchyma, and no distribution to the contralateral side. An alternative drug delivery strategy is implantation within the brain of either genetically engineered cells secreting a neurotrophin, or a polymeric matrix containing the neurotrophin. The coronal section autoradiogram was obtained 48 hours after the intracerebral implantation of a 2-mm diameter polymeric disc comprised of poly (ethylene-covinyl acetate) and 5 mg of [125I]-nerve growth factor (NGF) (Krewson et al., 1995). The NGF concentration immediately surrounding the polymeric implant is extremely high, but is decreased 10-fold within a diffusion distance of 500 μm from the polymer/tissue interface; the majority of the brain space containing NGF is occupied by the 2-mm polymeric disc. If the invasive strategies are unsuccessful, then it is usually advocated that small molecules provide a strategy for dealing with the brain drug delivery problem. However, even if a neurotrophin-mimetic small molecule was discovered, this drug would need a BBB drug delivery strategy if the small molecule mimetic lacks the dual criteria of lipid solubility and molecular weight less than a 400 to 600 d threshold (Fig. 2). Thus, in the absence of practical BBB drug delivery strategies, present day CNS drug discovery programs have a high likelihood of ending in program termination.
The fact that intraventricular drug administration only results in drug distribution to the ependymal surface is not at odds with the observation that the intraventricular administration of neuropeptides frequently results in pharmacologic effects in rats. The ICV injection of opioid peptides results in profound analgesia. However, this is because the site of action of the opioids is in the periaqueductal gray matter, which is immediately contiguous with the CSF flow tracks (Herz et al., 1970). The intraventricular administration of nerve growth factor (NGF) results in the sparing of medial forebrain cholinergic neuronal systems after fimbria fornix transection in rats (Koliatsos et al., 1991a) or primates (Koliatsos et al., 1991b), caused by the retrograde transport of NGF from axons terminating at the ependymal surface to areas deep within the brain (Seiler and Schwab, 1984; Ferguson et al., 1991).
In summary, intraventricular infusion is an ideal way of delivering drugs to the surface of the brain, but is a poor mode of delivering drugs into brain parenchyma. Second, the ICV injection of drug results in distribution to the ependymal surface of only the ipsilateral brain (Fig. 5), because of the unidirectional flow of CSF within the brain. Third, intraventricular administration is like a slow, intravenous infusion and the drug is readily distributed into the peripheral bloodstream after intraventricular drug infusion (Aird, 1984).
INTRACEREBRAL IMPLANTS
Neurotrophins may be locally released within the brain after the intracerebral implantation of either genetically engineered cells designed to secrete the neurotrophin (Kordower et al., 1994), or a polymeric implant that undergoes biodegradation with release of the neurotrophin (Krewson et al., 1995). After release of the neurotrophin from either the transplanted cell or the transplanted polymeric implant, the drug may penetrate into brain parenchyma only via diffusion, which decreases with the square of the diffusion distance, as predicted by the equation above. Because the weight of the human brain is a 1000-fold greater than the weight of the rat brain, the diffusion distances in the human brain are prohibitive for effective diffusion. The limited diffusion has been shown for both small molecules, such as 1,3-bis(2-chloroethyl)-1-nitrosourea, or for neurotrophins, such as NGF (Fig. 5). The brain concentration of 1,3-bis(2-choloroethyl)-1-nitrosourea is decreased 90% at a distance of 0.5 mm removed from the polymeric implant (Fung et al., 1996). There is effectively no measurable NGF in the brain at a distance of 0.5 mm removed from a polymeric implant that is itself 2 mm in diameter (Krewson and Saltzman, 1996). Given the limited efficacy of drug diffusion within the brain, it would be necessary to have multiple areas of the brain implanted with engineered cells or polymeric implants, because the effective treatment volume is <1 mm3 per implant. However, the cells immediately adjacent to the implant would be exposed to extremely high levels of the drug or neurotrophin.
BLOOD-BRAIN BARRIER DISRUPTION
The transient opening or disruption of the BBB after the intracarotid arterial administration of hypertonic solutions was first observed by Broman (1941) more than 50 years ago. In addition to hypertonicity, other membrane surface active agents may also cause BBB disruption including the intracarotid arterial infusion of low pH solutions (Nagy et al., 1985), oleic acid (Sztriha and Betz, 1991), cytochalasin B (Nag, 1995), or membrane alkylators, such as etoposide (Spigelman et, 1984), or melphalan (Cornford et, 1992). The intracarotid arterial infusion of hypertonic solutions has been applied in humans with brain tumors (Neuwelt et al., 1991). However, hyperosmolar BBB disruption causes opening of the BBB in the normal brain to a greater extent than BBB disruption in the tumor (Hiesiger et al., 1986). Moreover, the intracarotid arterial infusion of hypertonic solutions results in seizures in experimental animals (Neuwelt and Rapoport, 1984), as well as chronic neuropathologic changes (Salahuddin et al., 1988). The neuropathology after BBB disruption may be caused by the entry of albumin into brain interstitial space, because albumin is toxic to astrocytes (Nadal et al., 1995).
In addition to membrane surface active agents, vasoactive molecules also cause BBB disruption including prostaglandins (Schmidley et, 1992), histamine via an H2mechanism (Gross et al., 1981), C3a complement (Greenwood, 1991), bradykinin (Inamura and Black, 1994), 5-hydroxytryptamine (Winkler et al., 1995), and S-nitroso-acetylpenicillamine, a nitric oxide donor. The intracerebral injection of kainic acid also causes BBB disruption via an exitotoxic mechanism, and this disruption of the barrier may be attenuated by inhibitors of nitric oxide synthase (Mayhan and Didion, 1996). The bradykinin induced opening of the blood tumor barrier is also mediated via a nitric oxide mechanism (Nakano et al., 1996). Thus, vasoactive molecules that cause BBB disruption may have a final common pathway similar to exitotoxic brain damage, such as ischemia or hypoglycemia.
Whereas the disruption of the BBB in normal brain results in chronic neuropathologic sequelae (Salahuddin et al., 1988) and vascular damage (Lossinsky et al., 1995), the selective disruption of the BBB in brain tumors may have beneficial clinical effects by selectively delivering chemotherapeutic agents to the tumor, but not to the normal brain. The intracarotid arterial infusion of bradykinin results in the selective opening of the blood tumor barrier, but not the BBB in the normal brain (Inamura and Black, 1994). The administration of bradykinin is a form of biochemical BBB disruption that seems to selectively augment the permeability of a region of brain that already has a partially compromised BBB. The reason for the selectivity of bradykinin in opening the BBB in brain tumors, but not in the normal brain, may be that the bradykinin receptors are on the brain side of the BBB, and are not exposed to circulating bradykinin when the BBB is normally intact.
SMALL MOLECULES
Blood-brain barrier drug targeting strategies may not be needed if high throughput screening programs lead to “small molecules” that cross the BBB unassisted. However, this line of reasoning is problematical for two reasons. First, it is difficult to generate small molecule peptidomimetic drugs that retain high affinity binding and specificity for the target receptor. For example, erythropoeitin is a 165 amino acid peptide, and a 20 amino acid truncated form of erythropoeitin has been developed, which has a 20,000-fold decrease in receptor affinity (Wrighton et al., 1996). Secondly, a molecule is not small unless it has a molecular weight less than a 400 to 600 d threshold (Levin, 1980). Even then, a drug with a molecular weight under 500 d may not cross the BBB in pharmacologically active amounts unless the drug is lipid soluble. These dual criteria (lipid solubility, molecular weight less that 500 d) are unlikely to be fulfilled by small molecule peptido-mimetic drugs. For example, small molecule NGF-mimetics (LeSauteur et al., 1995) or neurotensin-mimetics (Cusack et al., 1995) are oligopeptide molecules that will still need BBB drug targeting vehicles to achieve CNS pharmacologic effects after systemic administration. A nonpeptide endothelin receptor antagonist has been developed (Ohlstein et al., 1994), but this compound, SB209670, has a molecular weight > 500 d and a dicarboxylic moiety that will prevent significant BBB transport in vivo. Therefore, even if a small molecule peptido-mimetic drug was discovered, this drug may still need to be conjugated to a BBB drug targeting system if CNS pharmacologic effects after systemic administration are to be achieved.
Molecular weight threshold for drug transport through the blood-brain barrier
The studies of Levin (1980) show that there is a molecular weight threshold of approximately 400 to 600 d with respect to drug penetration through the BBB. That is, BBB permeability is proportional to the lipid solubility of the drug, providing the molecular weight of the drug is under a 400 to 600 d threshold. Drugs with molecular weights above this threshold penetrate the BBB at rates much reduced from that predicted by the lipid solubility of the compound. For example, the BBB penetration of vincristine is reduced from the rate expected based on the high lipid solubility of the compound, because this drug has a molecular weight of 800 d (Grieg et al., 1990). Molecular weight thresholds of solute penetration through biological membranes are a general phenomenon (Hingson and Diamond, 1972; Xiang and Anderson, 1994). The mechanism of solute penetration through biological lipid bilayers may be pore-mediation. Trauble (1971) has suggested that transitory pores are formed in the lipid bilayer, and are caused by the “kinking” of the fatty acyl side chains that are in constant molecular motion within the biological membrane. The pore mechanism explains the molecular weight threshold of solute transport through biological membranes, and also explains the paradoxical observation of saturability of BBB transport of lipid soluble compounds (Pardridge, 1991).
P-glycoprotein
Many of the drugs identified by Levin (1980) as being poorly transported through the BBB may also be substrates for p-glycoprotein, the product of the multidrug resistance gene, which acts as a membrane active efflux system for a variety of drugs (Eytan et al., 1996). For example, vincristine is a substrate for p-glycoprotein (Pastan et al., 1991). The immunocytochemical detection of p-glycoprotein in brain microvessels (Cordon-Cardo et al., 1989; Jette et al., 1993), led to the hypothesis that p-glycoprotein is present at brain capillary endothelial membranes, specifically the endothelial luminal membrane (Stewart et al., 1996). In this context, brain endothelial luminal membrane p-glycoprotein is presumed to act as an active efflux system preventing drug transport across the BBB. However, the finding of immunoreactive antigen in brain microvessels is not an unequivocal demonstration that the antigen is of endothelial origin. This is because microvessel preparations also contain pericytes, smooth muscle, and astrocyte antigens. The surface of isolated brain microvessels is decorated with astrocyte foot process remnants that remain attached to the basement membrane after the brain capillary isolation procedure (White et al., 1981). Recent studies have used dual labeling confocal fluorescent microscopy to show that immunoreactive p-glycoprotein in human brain microvessels comigrates exactly with immunoreactive glial fibrillary acidic protein in these vessels (Pardridge et al., 1997). Conversely, microvascular p-glycoprotein did not colocalize with an endothelial antigen, the GLUT1 glucose transporter. These findings indicate that p-glycoprotein, at least with respect to human brain microvessels, is located at the microvasculature largely within the astrocyte foot processes. Although p-glycoprotein is found in brain capillary endothelial cells in tissue culture, the mdr1b gene is selectively up-regulated in tissue culture, whereas only the mdr1a gene is expressed in vivo (Barrand et al., 1995).
The localization of p-glycoprotein to astrocyte foot processes is not incongruous with the observation that brain/plasma ratios are increased in p-glycoprotein knockout mice for drugs that are substrates for p-glycoprotein (Schinkel et al., 1995). The brain/plasma drug ratios are volumes of distribution, which are a function of transport across astrocyte membranes and sequestration by cytoplasmic binding proteins, as well as BBB permeability. P-glycoprotein may normally serve to retard the uptake into astrocytes of drugs that initially cross the BBB.
Lipid soluble drug carriers and the pharmacokinetic rule
The attachment of water soluble drugs to lipid soluble carriers has been proposed as a strategy for targeting drugs through the BBB. Lipid carriers that have been used in the past include dihydropyridine (Bodor et al., 1981), adamantane (Tsuzuki et al., 1991), and fatty acyl carriers (Chekhonin et al., 1991), such as N-docosahexaenoyl (Shashoua and Hesse, 1996). The attachment of a lipid carrier to a drug may increase the lipid solubility of the compound, but the increased lipid solubility may not be translated into a proportional increase in BBB permeability. The latter is quantified by measurement of the brain capillary permeability-surface area product, which is also referred to as the Ki. The permeability-surface area product may not increase in proportion to lipid solubility if the molecular size of the drug/carrier complex is in excess of the 600 d threshold.
The principal problem with using lipid carriers is predicted by the pharmacokinetic rule (Fig. 6). The uptake of a drug in the brain, quantified as percent of injected dose delivered per gram of brain, is directly proportional to both the BBB permeability-surface area product and the plasma area under the concentration curve (AUC). When the lipid solubility of a drug is increased, the BBB permeability-surface area product may be increased, but the plasma AUC is decreased, generally in proportion to the increase in BBB permeability-surface area product. The decreased plasma AUC occurs because the enhanced lipid solubility of the compound results in an increased distribution of the drug into virtually all peripheral tissues with concomitant rapid removal from the bloodstream. Thus, the increased permeability-surface area product caused by the lipid carrier is offset by the decreased plasma AUC. For example, when azidothymidine is coupled to an adamantane carrier, the azidothymidine is removed from plasma 10 times faster relative to the unconjugated azidothymidine (Tsuzuki et al., 1994). Penetration of the drug into the CSF is actually decreased by attachment to the carrier because of the profound effect of the lipid carrier on the plasma AUC of the drug. The concentration of azidothymidine in brain tissue is unchanged after peripheral administration of free azidothymidine or azidothymidine in the form of the lipid carrier. This constancy of the percent injected dose per gram is caused by the offsetting effects of the increased permeability-surface area product and the decreased AUC caused by use of the lipid carrier. Apart from these pharmacokinetic considerations, the use of a lipid carrier may require that the drug still react with the target receptor in brain while the drug is conjugated to the lipid carrier. If the drug must be released in brain from the lipid carrier to interact effectively with the target receptor, then the covalent bond joining the drug and the lipid carrier must be cleaved faster than the drug/carrier complex is metabolized in brain.
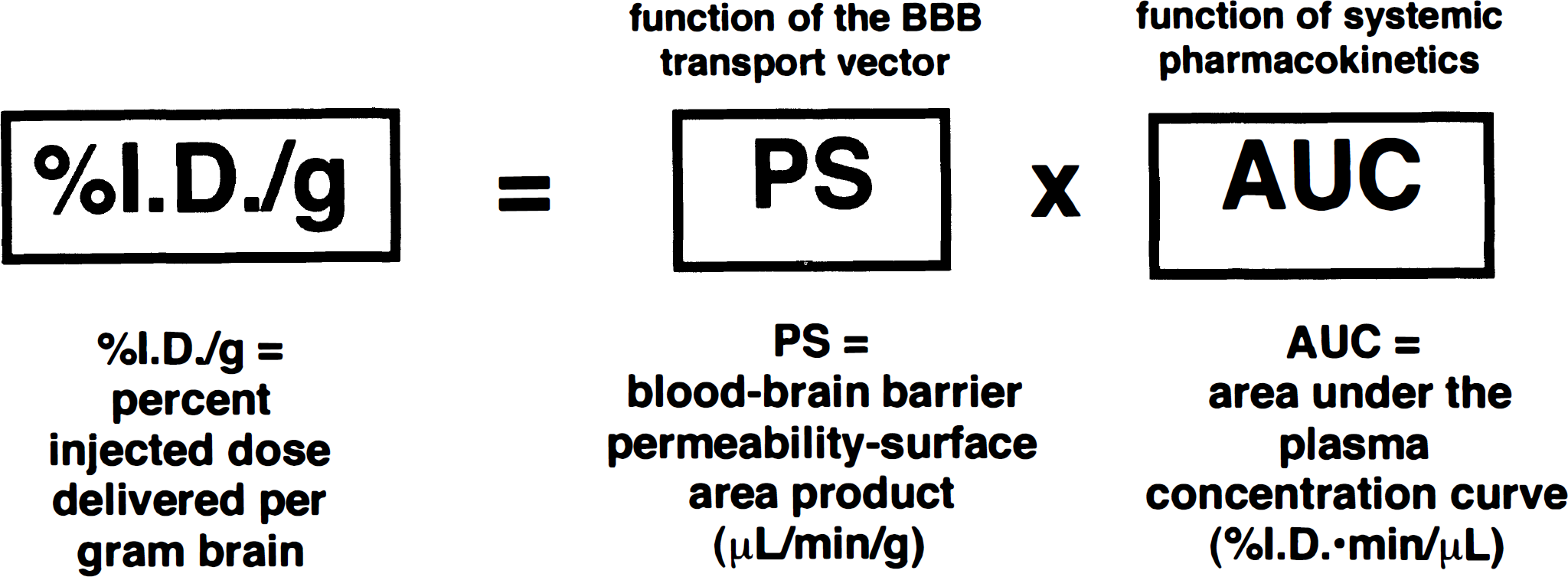
The pharmacokinetic rule for brain drug delivery states that the brain uptake, measured as percent of injected dose (ID) of drug delivered per gram of brain is a dual function of the BBB permeability-surface area product, and the plasma area under the concentration curve (AUC). The permeability-surface area product is a function of the BBB transport vector, and the plasma AUC is a function of the systemic pharmacokinetics of the drug, which is in turn a function of the intrinsic chemical properties of the drug. When small molecules are attached to lipid carriers, the BBB permeability-surface area product may increase several-fold. However, the organ clearance of the drug by peripheral tissues is also increased, which results in a proportional decrease in the plasma AUC. Thus, the increased permeability-surface area product and the decreased plasma AUC tend to have offsetting effects with minimal increases in percent ID/g being achieved with strategies that aim to increase the lipid solubility of the drug.
Summary
The current approach for brain drug delivery is exemplified in the case of the neurotrophins. In the early 1990s, there was considerable promise that these molecules would provide new treatments for multiple CNS disorders (Hefti and Schneider, 1991). This enthusiasm was generated by the rapid rate of progress in neurotrophin CNS drug discovery and the profound CNS effects that these drugs exerted when injected directly into the brain, bypassing the BBB. This CNS drug discovery evolved in the absence of significant progress in CNS drug delivery. Therefore, neurotrophin delivery to the CNS in phase I clinical trials was limited to the invasive strategies outlined in Fig. 5. The intraventricular infusion of neurotrophins was advocated even though preclinical autoradiographic studies showed that intraventricular neurotrophin is distributed only to the ipsilateral ependymal surface of brain (Fig. 4). An alternative strategy is intracerebral implants of either engineered cells or polymers infiltrated with neurotrophin. However, because the effective treatment volume around the implant is <1 mm3 (Fig. 5), this approach would require multiple implants within the brain to achieve a reasonable distribution of drug within the CNS. Moreover, the patients' multiple implants would require replacement at an interval of every 6 to 12 months. The backup strategy is the development of neurotrophin-mimetic “small molecules.” However, even if a neurotrophin-mimetic small molecule was discovered, this molecule would most likely require a BBB drug delivery strategy, because the chemical properties of the molecule would not fulfill the dual criteria of (a) lipid solubility, and (b) molecular weight < 400 to 600 d. Given the failure of the existing brain drug delivery strategies, termination of the CNS drug development program is inevitable (Fig. 5). Therefore, it is important to develop new forms of drug delivery to the brain that involve drug targeting through the BBB using normal physiologic transport processes. These include the multiple nutrient transporters and peptide receptors that normally function to carry across the BBB circulating nutrients or peptides via carrier-mediated and receptor-mediated transcytosis processes, respectively. The carrier-mediated transport of a drug through the BBB is reviewed in the context of pseudo-nutrients, and receptor-mediated transcytosis of drugs attached to transport vectors is reviewed in the context of chimeric peptides, pegylated immunoliposomes, and antisense therapeutics.
CARRIER-MEDIATED TRANSPORT
L-DOPA is the classical pseudo-nutrient that undergoes carrier-mediated transport through the BBB via the neutral amino acid transporter (Wade and Katzman, 1975). The delivery of drugs to the brain via BBB carrier-mediated transport is most likely to occur when the drug is modified to take on the structure of an endogenous nutrient. This approach is to be contrasted with the idea that a nontransportable drug could be covalently attached to a nutrient that acts as a carrier. The formation of a nutrient/drug conjugate would most likely induce structural changes within the nutrient beyond that tolerated by the stereospecific pore within the transporter protein. It is conceivable that some drug/nutrient conjugates could undergo carrier-mediated transport through the BBB, but the most likely event is carrier-mediated transport of a drug that has been modified such that the drug itself has a structure analogous to an endogenous nutrient. For example, L-DOPA has the structure of a neutral amino acid. Drugs that undergo carrier-mediated transport through the BBB other than L-DOPA include α-methyl-DOPA, melphalan, α-methyl-para-tyrosine, and gabapentin, which all undergo transport via the BBB neutral amino acid transport system. An example of converting the structure of a non-transportable drug into a pseudo-nutrient structure would be the case of a monoamine that normally does not undergo significant transport through the BBB. Rather than attach this drug to an amino acid, an alternative approach would be to convert the monoamine into an α-amino acid. This pro-drug would undergo transport through the BBB via the neutral amino acid carrier. Once in the brain, the pro-drug would be decarboxylated back to the parent monoamine via aromatic amino acid decarboxylase (Wade and Katzman, 1975).
RECEPTOR-MEDIATED TRANSPORT
Vector discovery
The brain capillary endothelium expresses specific receptors for circulating peptides or plasma proteins. Human brain microvessels have been used to characterize the human BBB receptors for insulin, insulin-like growth factor-I, insulin-like growth factor-II, transferrin, and leptin (Fig. 7). Brain microvessels also express a receptor for low-density lipoprotein (Dehouck et al., 1994), which is separate from the scavenger receptor that is responsible for the receptor-mediated endocytosis of modified low-density lipoprotein such as acetyl low-density lipoprotein (Triguero et al., 1990). There are also absorptive-mediated endocytosis systems at the brain capillary endothelium that recognize either the carbohydrate or cationic charge of the protein (Fig. 7). The BBB insulin, insulin-like growth factor, and transferrin receptor systems have been shown to mediate the transcytosis of these peptides through the BBB in vivo (Duffy and Pardridge, 1987; Fishman et al., 1987; Rheinhardt and Bondy, 1994). The observation that peptide receptors are present on brain capillary endothelium, and that some of these mediate peptide transcytosis through the BBB led to the chimeric peptide hypothesis (Pardridge, 1991). Chimeric peptides are formed when a nontransportable drug is covalently attached to a peptide or protein that normally undergoes receptor-mediated transcytosis through the BBB (Fig. 7). The latter acts as a vector for drug targeting through the BBB. The use of insulin, per se, as a vector would be problematical because the administration of the drug/insulin conjugate would cause hypoglycemia. The use of transferrin itself as a drug delivery vector is prevented by the large concentration of transferrin in the plasma, which is about 25 μM, a level that is 3 log orders of magnitude in excess of the dissociation constant (KD) governing the BBB transferrin receptor/transferrin binding reaction (Pardridge et al., 1987). Therefore, the initial vector used for the purposes of delivery across the BBB was cationized albumin which undergoes absorptive-mediated transcytosis through the BBB (Kumagai et al., 1987). Subsequently, the OX26 murine monoclonal antibody (MAb) to the rat transferrin receptor was developed (Friden et al., 1993), and this MAb has an approximately fourfold greater BBB permeability as compared with cationized albumin (Pardridge, 1995). The most active BBB transport vector discovered to date is the 83–14 murine monoclonal antibody to the human insulin receptor, which has a BBB permeability in the primate of 5.4 ± 0.6 μL/min/g (Pardridge et al., 1995). This permeability-surface area product is ninefold greater than BBB permeability-surface area product for an antitransferrin receptor MAb in either the rat (Pardridge, 1995) or the primate (Walus et al., 1996).
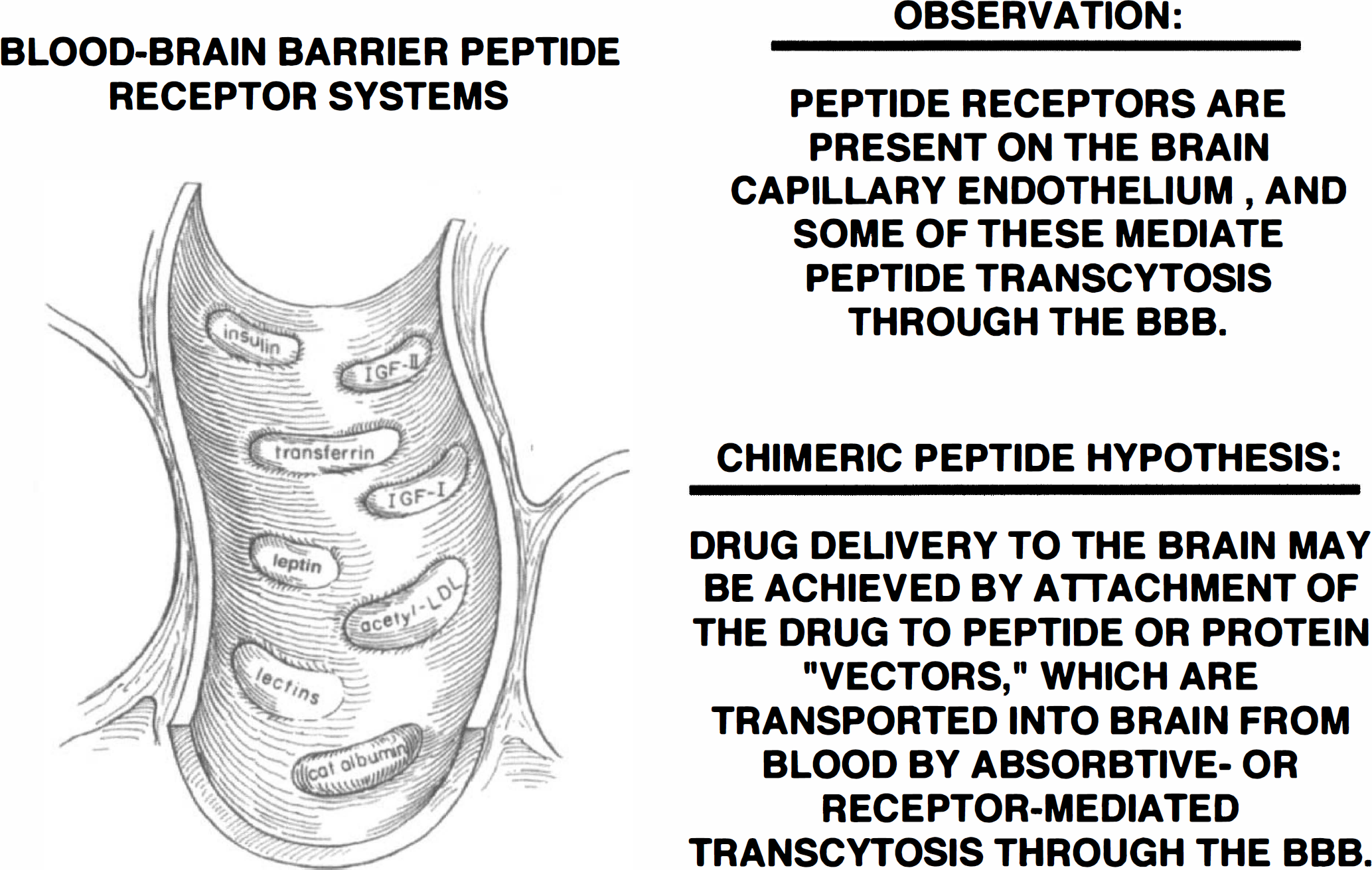
Blood-brain barrier peptide receptor systems include those for insulin, insulin-like growth factor (IGF)-I, IGF-II, transferrin, and leptin (Golden et al., 1997). The insulin, IGF, transferrin, and leptin receptor systems are believed to mediate the transcytosis of these circulating ligands through the brain capillary endothelium and into the brain interstitial fluid. The scavenger receptor binds modified proteins such as acetylated low density lipoprotein and mediates only the endocytosis of acetylated low-density lipoprotein (LDL) into the brain capillary endothelium, not transcytosis (Triguero et al., 1990). Lectins, such as wheat germ agglutinin, bind to brain endothelial carbohydrate sites (Broadwell et al., 1988), and cationic proteins, which bind to brain endothelial anionic sites, undergo absorptive-mediated transcytosis through the BBB (Triguero et al., 1990).
The 83–14 MAb binds to an epitope within amino acids 469 to 592 of the α subunit of the human insulin receptor (Prigent et al., 1990), as depicted in Fig. 8A. This epitope projects into the extracellular space, i.e. the plasma compartment of brain, and is freely accessible to the circulating MAb. The 83–14 MAb strongly illuminates the capillary endothelium in sections of Old World primate (rhesus monkey) brain (Fig. 8B), but does not react with microvessels in brain of New World primates, such as the squirrel monkey. This correlates with the closer genetic similarities between humans and Old World primates, as opposed to New World primates (Swindler and Erwin, 1986). The distribution of [125I]-MAb 83–14 into the brain of rhesus monkeys in vivo is extraordinarily high and the brain volume of distribution reaches 1.3 ml/g, a volume of distribution in excess of the glucose brain volume of distribution (Fig. 8C).
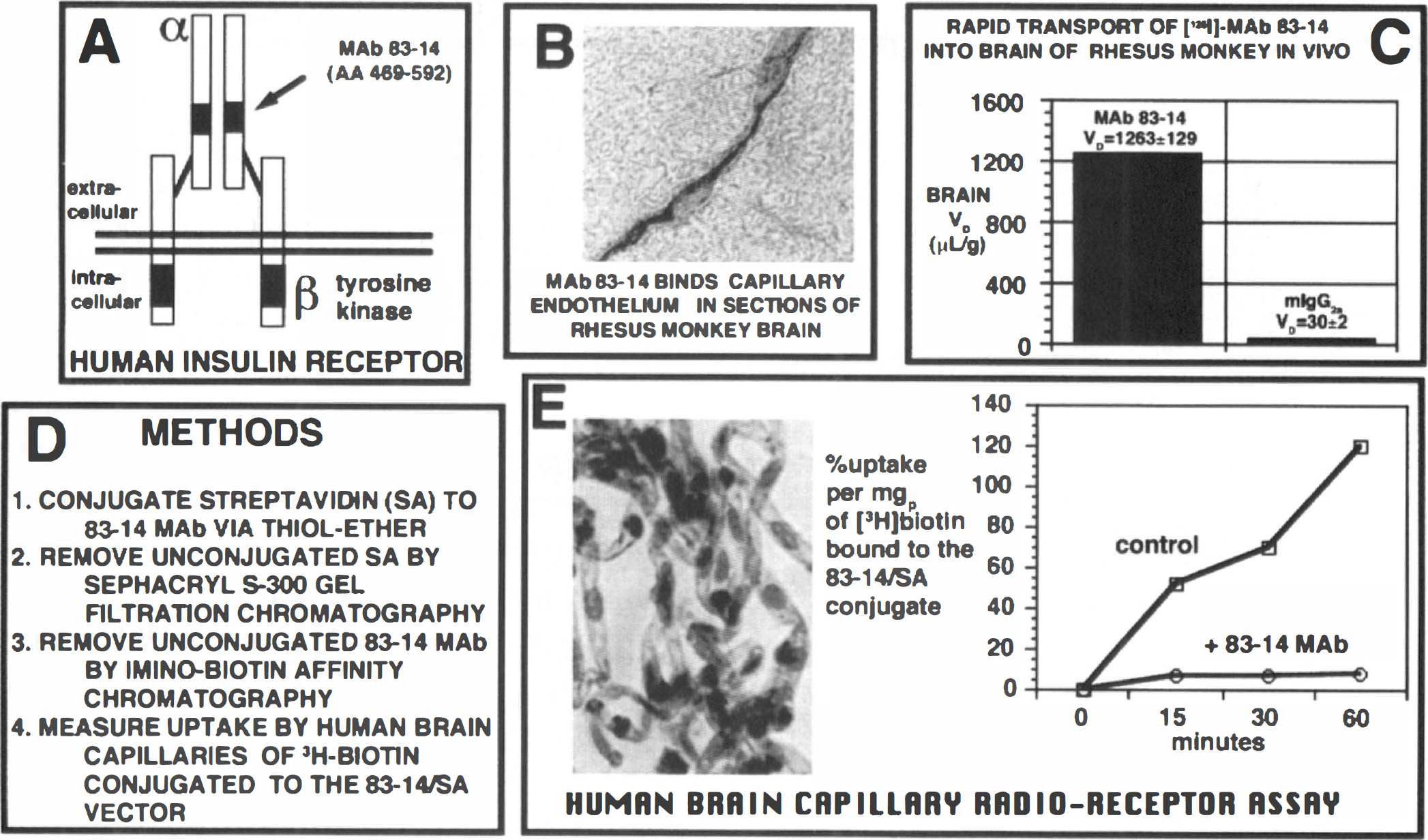
Drug targeting to primate or human brain via a monoclonal antibody (MAb) to the human insulin receptor.
Conjugation strategy based on avidin-biotin technology
The 83–14 MAb may be converted into a universal brain drug delivery system that could potentially transport across the BBB any biotinylated substance by first conjugating streptavidin (SA) to the MAb via a stable thiolether linker (Fig. 8D). The unconjugated streptavidin was removed by Sephacryl S300 gel filtration, and the unconjugated 83–14 MAb was removed by iminobiotin affinity chromatography. The bifunctionality of the 83–14/SA conjugate was assayed using human brain capillaries and the measurement of [3H]-biotin uptake (Fig. 8E). The uptake of the [3H]-biotin/SA-8314 conjugate by isolated human brain capillaries was high and was completely suppressed by unconjugated 83–14 MAb (Wu et al., 1997).
Monoclonal antibody/avidin fusion proteins have been produced with recombinant DNA technology from an MAb/avidin fusion gene as recently described (Shin et al., 1997). The plasma pharmacokinetics approximate the MAb, not avidin (Shin et al., 1997). The immunogenicity of the murine MAb component of an MAb/avidin fusion protein in humans may be eliminated by “humanization” of the murine framework sequences by genetic engineering. The lack of significant antigenicity of avidin in humans (Paganelli et al., 1994) may be caused by the high concentration of avidin in the diet, which can induce tolerance through oral antigen feeding (Weiner, 1994).
Experimental studies of MAb/avidin conjugates use the neutral analogue, streptavidin, not the cationic avidin, because the use of streptavidin optimizes plasma pharmacokinetics of Mab-avidin complexes that are conjugated by chemical linkers (Pardridge, 1995). The availability of the 83–14/SA conjugate allows for delivery of biotinylated drugs through the BBB in Old World primates or humans in vivo, and the availability of the OX26/SA conjugate allows for brain drug targeting in rats, as these MAb are species-specific. The OX26 vector has been used for brain drug targeting of peptide-based therapeutics such as a vasoactive intestinal peptide (VIP) analog, neurotrophins, and antisense agents including phosphorothioate oligodeoxynucleotides (PS-ODN) and peptide nucleic acids (PNA). The 83–14 MAb has been used to develop a peptide radiopharmaceutical for the semi-quantitation of the A[beta] amyloid burden in the human brain with Alzheimer's disease.
Vasoactive intestinal peptide chimeric peptides
Vasoactive intestinal peptide causes cerebrovasodilatation when directly applied to pial vessels (Suzuki et al., 1984), but there is no enhancement of cerebral blood flow when the neuropeptide is infused directly into the carotid artery (McCulloch and Edvinsson, 1980). This is because VIP does not normally undergo transport through the BBB (Bickel et al., 1993). A VIP therapeutic was developed by specifically designing a VIP analog to allow for monobiotinylation. The multibiotinylation of VIP would lead to the formation of high molecular weight aggregates after conjugation to MAb/avidin vectors, because of the multivalency of avidin binding of biotin. The VIP analog was designed for monobiotinylation by replacing lysine residues with arginine residues and acetylating the amino terminus of the peptide (Bickel et al., 1993). After biotinylation, the VIP therapeutic was attached to the OX26/SA vector and injected intravenously into conscious rats (Wu and Pardridge, 1996). This resulted in a 66% increase in whole brain blood flow at a relatively low systemic dose of 20 μg/kg of neuropeptide. In the absence of the BBB drug targeting system, the VIP analog caused no change in cerebral blood flow. Conversely, the intravenous injection of the unconjugated VIP analog caused a 350% increase in salivary gland blood flow caused by the rapid transport of the VIP through the large pore system within salivary gland capillaries (Wu and Pardridge, 1996). However, the attachment of the VIP to the OX26/SA vector completely inhibited the increase in salivary gland blood flow, because the VIP/SA-OX26 conjugate was too large to undergo transport through salivary gland capillary pores. Therefore, attachment of the VIP to the OX26/SA vector resulted in organ-specific drug targeting that caused both a CNS pharmacologic effect and a parallel suppression of the pharmacologic effect of the neuropeptide in peripheral tissues. The therapeutic index of the VIP therapeutic was increased more than 1,000% by attachment to the BBB drug delivery system (Wu and Pardridge, 1996).
Neurotrophin chimeric peptides
Neurotrophins such as NGF or brain-derived neurotrophic factor have been attached to the OX26 vector and this resulted in a measurable brain delivery of the neurotrophin (Friden et al., 1993; Granholm et al., 1994; Pardridge et al., 1994). In contrast, the intravenous administration of the unconjugated neurotrophin resulted in no neurotrophin distribution to the brain because of lack of transport through the BBB (Pardridge et al., 1994). The increase in brain uptake of the NGF or brain-derived neurotrophic factor is relatively modest. For example, the maximal brain uptake of NGF/OX26 chimeric peptide was on the order of 0.03% of injected dose per gram brain (Granholm et al., 1994), a value that is more than 10-fold reduced compared to the maximal brain uptake of the unconjugated OX26 MAb, which is on the order of 0.4% injected dose per gram brain in the rat (Pardridge, 1995). The 10-fold reduction in brain uptake of the NGF/OX26 conjugate relative to OX26 alone is caused by a pharmacokinetic problem that is intrinsic to the NGF-like neurotrophins. These proteins are all strongly cationic and are rapidly removed from the bloodstream because of rapid uptake by the liver (Pardridge et al., 1994). The neurotrophins provide an example of how unfavorable pharmacokinetic properties cause limitation in terms of brain delivery of drugs. A very active BBB transport vector may be available, but if the intrinsic properties of the drug are such that the pharmacokinetics are unfavorable, and there is rapid removal of the drug from the bloodstream, then the maximal brain delivery of the drug with the use of the BBB transport vector is attenuated. In this case, it is necessary to focus on strategies for both (1) enhancement of BBB transport via attachment to BBB transport vectors (permeability surface area, Fig. 6), and (2) optimization of the plasma pharmacokinetics (AUC, Fig. 6).
Neurotrophin pegylation
Optimization of protein pharmacokinetics may be accomplished by protein pegylation, which involves attachment of 2,000 to 5,000 d polyethyleneglycol polymers to surface amino groups on the protein (Nucci et al., 1991). However, the conjugation of free amino groups of NGF-like neurotrophins results in a loss of biologic activity of the protein (Rosenberg et al., 1986). An alternative strategy that has been recently developed is to pegylate the neurotrophin via carboxyl moieties of glutamate or aspartate residues (Sakane and Pardridge, 1997). The carboxyl-directed pegylation of brain-derived neurotrophic factor results in a profound slowing of the rate of clearance of the neurotrophin from the plasma compartment and an optimization of the plasma pharmacokinetics of the neurotrophin. The use of bifunctional polyethyleneglycol moieties allows for facile conjugation of biotinylated/pegylated neurotrophins to MAb/avidin BBB transport vectors.
Peptide radiopharmaceuticals
In addition to peptide-based therapeutics, it is also possible to deliver peptide radiopharmaceuticals through the BBB for the purposes of diagnosing brain disorders. For example, brain tumors that selectively express the epidermal growth factor receptor could be detected by the BBB delivery of a radiolabeled epidermal growth factor peptide radiopharmaceutical. The amyloid burden in the brain of patients with Alzheimer's Disease may be semi-quantitated by delivering across the BBB an amyloid imaging agent. The amyloid in the brain of the patient with Alzheimer's Disease is caused by the deposition of a 43 amino acid A[beta] amyloidotic peptide. The first 40 amino acids of this peptide, designated A[beta]1–40, is soluble in aqueous solution and may be iodinated. The [125I]-A[beta]1–40 deposits at pre-existing amyloid in sections of brains with Alzheimer's Disease (Maggio et al., 1992), and is a more sensitive marker of tissue amyloid than is a antibody directed against an amyloidotic component. However [125I]-A[beta]1–40 undergoes negligible transport through the BBB (Saito et al., 1995), because there is no specific receptor for this neuropeptide at the BBB. A[beta]1–40 has been iodinated and biotinylated and conjugated to the OX26/SA delivery system. As shown is Fig. 9A, this amyloid imaging agent is comprised of three domains: an amyloid binding domain consisting of the radiolabeled A[beta]1–40; a linker domain consisting of a 14-atom spacer attached to biotin and streptavidin; and a BBB transport domain consisting of the anti-receptor MAb that may either be the OX26 MAb for studies in rats or the 83–14 MAb for studies in Old World primates or humans. The A[beta]1–40 still binds to the amyloid in sections from the autopsy of a brain from a patient with Alzheimer's Disease despite conjugation to the 200,000 d OX26/SA transport vector, as shown by film autoradiography with sections of a brain from a patient with Alzheimer's Disease (Fig. 9B).
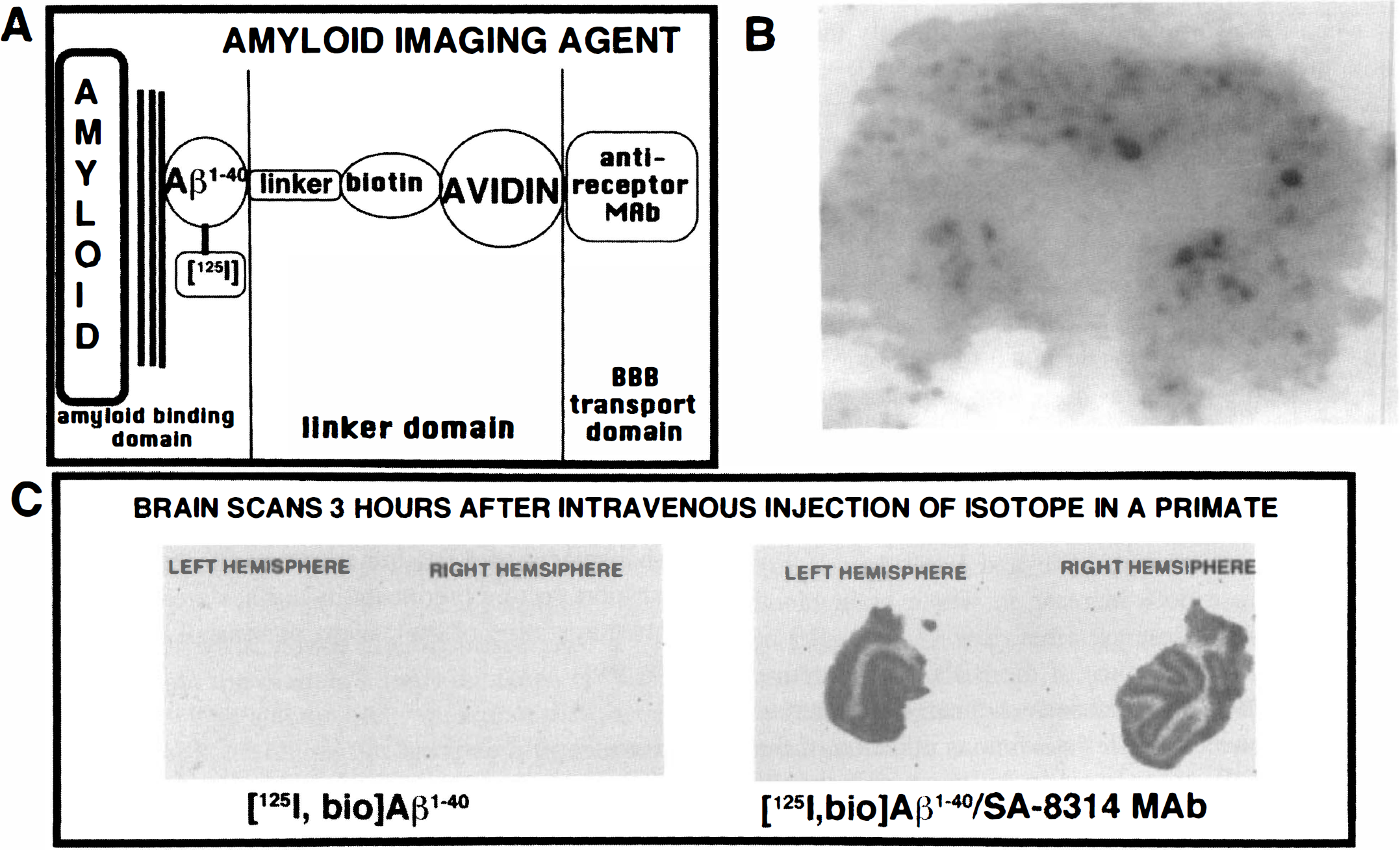
The amyloid imaging agent is comprised of three domains: radiolabeled A[beta]1–40 that binds to the amyloid of a brain with Alzheimer's Disease (AD); the linker domain which is comprised of a 14-atom spacer attached to a biotin moiety that is bound to an avidin analog such as streptavidin; a BBB transport domain comprised of an anti-receptor MAb such as the 83-14 MAb to the human BBB insulin receptor for studies in Old World primates or humans, or the OX26 MAb to the rat transferrin receptor for studies in rats (Saito et al., 1995).
As a next step toward developing an in vivo amyloid imaging agent in humans, the [125I,biotinyl]A[beta]1–40 was conjugated to the 8314/SA delivery system and injected intravenously into young anesthetized rhesus monkeys that lack CNS amyloid (Wu et al., 1997). When the A[beta]1–40 peptide was injected without the use of the 83–14 BBB drug targeting system, there was no measurable brain uptake of the neuropeptide at 3 hours after intravenous administration in rhesus monkeys (Fig. 9C, left panel). Conversely, when the A[beta]1–40 was conjugated to the 8314/SA BBB delivery system, there was a profound increase in the brain uptake and the image was comparable to a “2-deoxyglucose” scan, wherein regions of gray and white matter could be precisely delineated (Fig. 9C, right panel). Fig. 9C is a visual representation of the fact that it is now possible to target to the brain drugs that normally do not undergo significant transport through the BBB. The use of BBB drug targeting systems such as the 83–14 MAb allows for the application of peptide based pharmaceuticals as both therapeutic and diagnostic agents for the CNS in humans.
SMALL MOLECULE DELIVERY TO THE BRAIN USING PEGYLATED IMMUNOLIPOSOMES
Small molecules may be delivered thorugh the BBB by conjugation to delivery vectors, such as the 83–14 MAb. However, the number of small molecules that can be individually conjugated to MAb vectors is limited. The carrying capacity of the vector could be greatly expanded by attaching liposomes to the vector, because up to 10,000 small molecules can be sequestered within a single liposome. Liposomes are not normally transported across the BBB because these devices are too large to undergo lipid-mediated transport across the endothelial membrane (Gennuso et al., 1993). Even small unilamellar vesicles on the order of 40 to 80 nm do not accumulate in brain; multilamellar vesicles accumulate in brain because these particles (0.3 to 2 μm) cause embolism of brain capillaries (Schackert et al., 1989). If liposomes are directly injected into the brain, these particles dissolve in the lipid membranes of brain cells. Methyl-bicucculline liposomes cause seizures after intracerebral injection (Tokes et al., 1980). No seizures are observed after intravenous injection of the liposomes because of the failure of these particles to undergo transport through the BBB.
The attachment of delivery vectors, such as the OX26 or 83-14 MAb, to the surface of liposomes could mediate the transport of these small unilamellar vesicles through the BBB via a receptor-mediated transcytosis. However, liposomes (with or without MAb attached) are rapidly removed from the bloodstream via uptake by cells lining the reticulo-endothelial system (Chonn et al., 1992). Because of this rapid removal, the plasma AUC of liposomes is markedly reduced, and this results in unfavorable pharmacokinetics (Fig. 6). The rapid removal of liposomes from plasma may be inhibited by attachment of polyethyleneglycol polymers to the surface of the liposomes (Papahadjopoulos et al., 1991). However, this attachment would interfere with the receptor binding of MAb that are also attached to the surface of the liposome. The dual objectives of optimizing both BBB permeability, via vector-mediated drug delivery across the BBB, and optimizing plasma pharmacokinetics, via the use of pegylation technology, may be achieved by tethering the receptor-specific MAb to the tip of the polyethyleneglycol tail (Huwyler et al., 1996). That is, the 2,000 to 5,000 d polyethyleneglycol acts as a bridge between the liposome and the MAb that targets the complex through the BBB (Fig. 10A). The construction of such a complex is made possible by preparation of a bifunctional polyethyleneglycol derivative containing a phospholipid at one end and a maleimide group at the other end, which allows for formation of a stable thiolether linkage after conjugation with thiolated MAb (Fig. 10A).
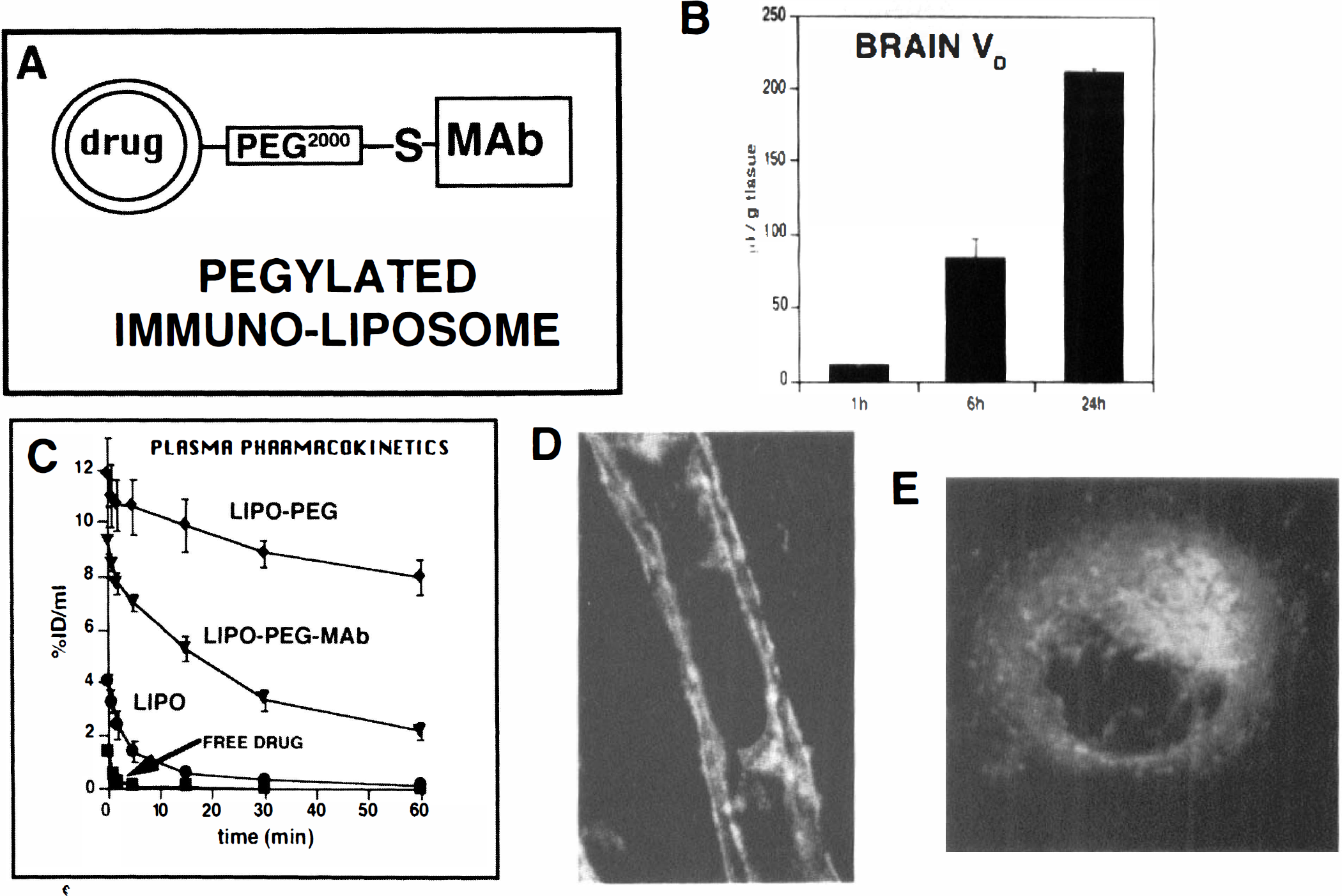
Strategy for conjugating a receptor-specific MAb to a liposome via tethering of the MAb via a stable thiol-ether (S) linkage at the tip of a polyethyleneglycol (PEG) tail that is in turn linked to a phospholipid within the surface of the liposome (Huwyler et al., 1996).
[3H]-daunomycin was encapsulated within liposomes and injected intravenously into rats. The brain volume of distribution (VD) of the [3H]-daunomycin increased with time indicating BBB transport and brain sequestration of the daunomycin entrapped in the pegylated immunoliposome (Fig. 10B). The plasma pharmacokinetics of the pegylated immunoliposome were optimized compared to nonpegylated liposomes. As shown in Fig. 10C, [3H]-daunomycin injected as free drug is removed extremely rapidly from the bloodstream after intravenous administration. Entrapment of the [3H]-daunomycin in conventional (i.e., nonpegylated) liposomes (designated LIPO in Fig. 10C) results in only a marginally increased plasma AUC compared to free drug (Fig. 10C). Conversely, the pegylation of the liposome (designated LIPO-PEG in Fig. 10C) results in a marked slowing in the rate of plasma clearance of the [3H]-daunomycin entrapped in the pegylated liposome. The rate of removal of the pegylated liposome is subsequently increased by tethering the receptor-specific MAb to the tip of the polyethyleneglycol tail (Fig. 10C). However, the rate of removal of the OX26 pegylated immunoliposome, 0.91 ± 0.11 mL/min/kg, is 14-fold slower than the rate of removal of conventional (nonpegylated) liposomes, 12.6 ± 6.3 mL/min/kg (Huwyler et al., 1996).
The OX26 MAb tethered to the pegylated immunoliposome still binds to the rat transferrin receptor and is also able to mediate the endocytosis of the complex into cells as shown by the confocal microscopy studies in panels D and E of Fig. 10. In these studies, a rhodamine-conjugated phospholipid was incorporated into the immunoliposomes, and the OX26 pegylated immunoliposomes were incubated with freshly isolated rat brain capillaries, or rat glioma (RG)-2 cells in tissue culture (panels D and E, respectively, Fig. 10). The confocal microscopy shows that the OX26 pegylated immunoliposome reacts with both luminal and abluminal membranes on brain capillary endothelium, indicating transferrin receptor is present on both membranes (Fig. 10D). Similarly, transferrin receptor is expressed on both apical and basolateral membranes of MDCK cells, which also perform receptor-mediated transcytosis of transferrin (Odorizzi et al., 1996). The OX26-pegylated immunoliposomes undergo endocytosis into RG2 cells in tissue culture (Fig. 10E). The average diameter of the liposomes used in these studies was measured by quasielastic light scattering, and was 85 nm (Huwyler et al., 1996). This approximates the size of brain capillary transcytotic vesicles, which have been measured by electron microscopy. In these studies, a 10 nm gold/OX26 conjugate was infused into the internal carotid artery of anesthetized rats for 10 minutes followed by fixation and embedding of brain tissue, and viewing under the electron microscope (Bickel et al., 1994). These studies provided evidence for transcytosis of the OX26/gold conjugate through the brain capillary endothelial compartment via endosomal structures that have an average diameter of approximately 100 nm.
ANTISENSE DRUG DELIVERY TO THE BRAIN
Antisense oligodeoxynucleotides (ODN) are potentially powerful CNS therapeutics that may inactivate translation of specific transcripts or facilitate premature degradation of specific transcripts via RNase H activity (Stein and Cheng, 1993). Phosphodiester (PO)-ODN are too labile in either blood or brain to be effective pharmaceuticals because of the rapid inactivation of PO-ODN by 3′-exonucleases (Fig. 11). Phosphorothioate (PS)-ODN are metabolically stable and resistant to 3′-exonucleases. However, PS-ODN bind nonspecifically to a variety of cellular proteins, and this causes nonsequence-related pharmacologic effects. For example, PS-ODN inhibit HIV uptake in lymphocytes by nonspecifically binding to CD4 and interfering with viral transport across the cell membrane, and this effect is unrelated to the specific nucleotide sequence of the antisense agent (Yakubov et al., 1993). Although PS-ODN are nonspecifically taken up by cells in culture, there is no measurable brain uptake of PS-ODN after systemic administration (Wu et al., 1996). Accordingly, PS-ODN have been delivered to the brain by intraventricular infusion. However, if the infusion dose generates an intrathecal concentration greater than 1 μM PS-ODN, then severe neurotoxicity ensues (Fig. 11), and can include cord necrosis, paralysis, and death (Whitesell et al., 1993; Wojcik et al., 1996).
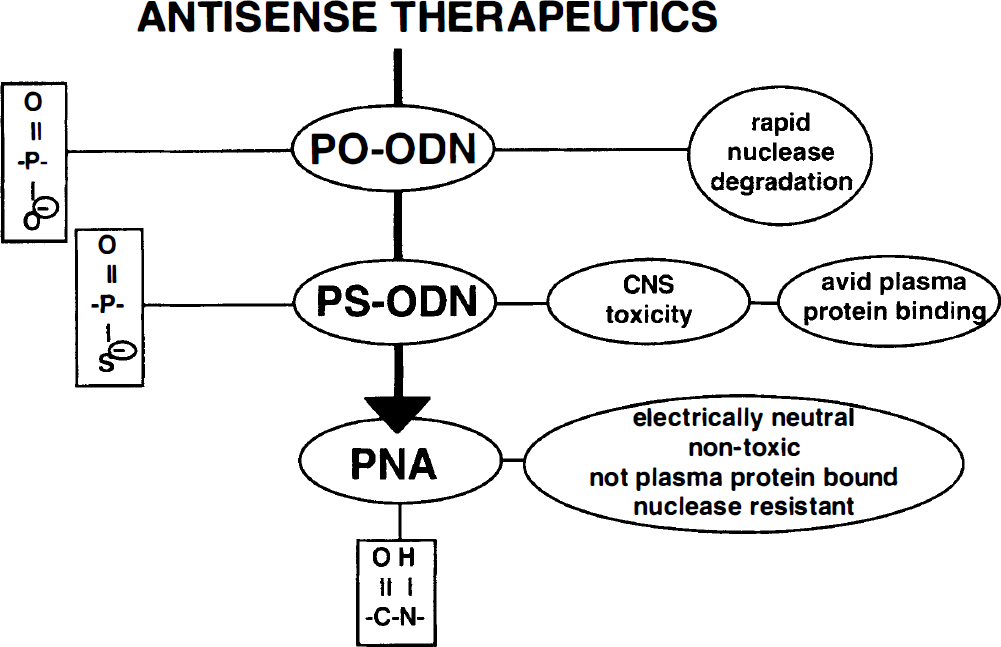
Antisense therapeutics include phosphodiester (PO) oligodeoxynucleotides (ODN), phosphorothioate (PS) ODN, and peptide nucleic acids (PNA). The PO-ODN and PS-ODN are polyanionic, but the PNA have an electrically neutral peptide backbone. The PNA lack the nuclease susceptibility of the PO-ODN, and the CNS toxicity or plasma protein binding problems of the PS-ODN.
The susceptibility of PO-ODN to 3′-exonuclease may be eliminated by conjugation of biotin (bio) to the 3′-terminus of the PO-ODN, which also allows for conjugation to an avidin-based BBB delivery system (Boado and Pardridge, 1992). However, when the 3′-bio-PO-ODN conjugated to the OX26/SA vector was injected intravenously into rats, rapid degradation of the ODN complex occurred, because of active endonuclease activity in vivo (Kang et al., 1995). This endonuclease activity is not present in cells in tissue culture (Boado and Pardridge, 1994), but is a major factor to be considered in the design of PO-ODN for in vivo applications. It is generally believed that the advantages of both PO-ODN and PS-ODN may be partially retained by the preparation of PO/PS-ODN hybrids. In this case, the internucleotide bond alternates between PO and PS linkages. However, when a PS-ODN is designed with a single, internal PO linkage, there is still rapid degradation of the antisense agent in vivo because of endonucleolytic attack of the single, internal PO linkage (Boado et al., 1995).
Blood-brain barrier transport of PS-ODN is negligible in the absence of drug targeting systems (Wu et al., 1996). However, the brain uptake of a 3′-bio-PS-ODN was increased to a level comparable to the brain uptake of morphine after conjugation of the 3′-bio-PS-ODN to the OX26/SA delivery system and intracarotid arterial infusion of the conjugate (Wu et al., 1996a). The 3′-bio-PS-ODN conjugated to the OX26/SA vector was then injected intravenously in rats, and a 20-fold reduction in BBB permeability-surface area product was observed because of severe plasma protein binding effects. Phosphorothioate-ODN are avidly bound by plasma proteins, such as albumin and α2-macroglobulin. The latter is a 700,000 d protein, and the binding of this very large plasma protein to the PS-ODN greatly restricted the BBB transport of the 3′-bio-PS-ODN conjugated to the OX26/ SA vector (Wu et al., 1996).
The disadvantages of PO-ODN (i.e., metabolic instability), or PS-ODN (i.e., neurotoxicity, plasma protein binding, and sequence unrelated nonspecific effects), can be eliminated by the use of PNA as the antisense agent (Fig. 11). A monobiotinylated model 18-mer PNA was conjugated to the OX26/SA vector and injected intravenously into rats (Pardridge et al., 1995). The brain uptake of the unconjugated PNA was negligible after intravenous administration, but the brain uptake of the PNA was increased 28-fold when the bio-PNA was conjugated to the OX26/SA vector, and this brain uptake approximated that of morphine. The PNA bound to the OX26/SA vector retained the ability to bind to the target mRNA. In this case, the model PNA was an 18-mer that was antisense to the rev gene of human HIV. The rev mRNA was prepared from a transcription plasmid, and was used in RNase protection assays. These studies showed that the PNA still bound to the target mRNA in a sequence-specific mechanism despite conjugation to the OX26/SA vector (Pardridge et al., 1995). In summary, the transport of unconjugated PNA across the BBB is negligible, but brain delivery of these neuropharmaceuticals can be achieved by coupling to vector-mediated BBB drug delivery systems.
3-Membrane barrier for antisense drug targeting
The problem with antisense drug delivery to the brain is that the target mRNA molecules reside in the cytoplasmic compartment of brain cells. Therefore, to exert a specific antisense effect, the molecule must undergo transport across three membranes in a series: the BBB, the brain cell plasma membrane, and the endosomal membrane. The OX26 MAb or the 83-14 MAb may mediate transport across the first two membranes, but cannot mediate endosomal release of the antisense agent. Therefore, targeting systems in the future that mediate brain delivery of antisense therapeutics must expand the multifunctionality of the targeting system to allow for not only transport through the BBB and endocytosis into brain cells, but also for drug release from the endosomal compartment.
Gene delivery to the brain
The expression of trans-genes in brain has been achieved with invasive methodologies that either by-pass the BBB (Freese et al., 1990; Le Gal La Salle et al., 1993; Suhr and Gage, 1993) or cause BBB disruption (Doran et al., 1995). Because receptor-mediated transcytosis of pegylated immunoliposomes through the BBB is possible (Fig. 10), and cationic liposomes entrap DNA with high efficiency (Felgner et al., 1994), the delivery of genes through the BBB with cationic liposomes might be envisaged. However, the cationic liposome/DNA complexes form aggregates in physiological saline (Wong et al., 1996), and the mechanism of uptake of these particles by cells in tissue culture is phagocytosis, not endocytosis (Matsui et al., 1997). If cationic liposome/DNA particles do form aggregates on entry into the bloodstream, then microembolization in the pulmonary circulation would be expected. In fact, >95% of the injected dose of a cationic liposome/DNA complex is removed by the lung on the first circulatory passage (Osaka et al., 1996), and the expression of the transgene in lung is 2 to 3 log orders greater than gene expression in liver after intravenous injection (Osaka et al., 1996; Liu et al., 1997). No gene expression in the CNS has been observed after the intravenous administration of cationic liposome/DNA complexes (Osaka et al., 1996), presumably because so little of the complex reaches the systemic circulation.
CONCLUSION
Brain drug targeting through the BBB must be achieved if present day CNS drug discovery platforms are to yield effective pharmaceuticals in the clinic. It is unlikely that the millions of patients afflicted with CNS disorders will tolerate, in a sustained way, the repetitive use of craniotomy to deliver drugs behind the BBB. What is needed is the development of BBB drug targeting systems that may be used by patients at home on a daily basis, involving no more that conventional subcutaneous administration of the delivery agent attached to the CNS pharmaceutical. For the pace of innovation in CNS drug delivery to quicken, the field of brain drug delivery must merge with CNS drug discovery as early in the overall CNS drug development process as possible. The development of safe and effective BBB drug targeting strategies is as complex and difficult as CNS drug discovery. However, technologies are available for continued innovation in the field, and future research should yield newer generations of brain-specific vectors, neuron-specific targeting, and endosomal release systems.