
Abstract

As discussed in the comprehensive and stimulating hypothesis article by Paschen and Doutheil (1999), neuronal cell damage in acute neurodegenerative disease traditionally has been linked to a perturbed cell calcium metabolism (for a recent review article, see Kristiàn and Siesjö 1998). Central to the calcium hypothesis of cell death is the fact that a compromise of the bioenergetic status of the cell leads to an increase in the free cytosolic calcium concentration (Ca2+i). This increase is caused by an influx of extracellular Ca2+ through plasma membrane channels modulated by voltage or agonists, particularly that gated when glutamate activates N-methyl-D-aspartate receptors, and to release of calcium ions from intracellular stores and binding sites. The major stores are endoplasmic reticulum (ER) and less well characterized intracellular organelles (“calciosomes”) which release calcium in response to an increase in Ca2+i (Ca2+-dependent Ca2+ release). The mitochondria can also release Ca2+, particularly if they have previously been loaded with calcium, e.g. by intense cellular activity or during reperfusion after ischemia.
Once Ca2+i increases to nonphysiologic levels, a host of potentially harmful calcium-dependent enzymes are activated. These encompass phospholipases, proteases, and endonucleases, as well as the neuronal and endothelial forms of nitric oxide synthase. However, as recalled by Paschen and Doutheil (1999), calcium-dependent cell death may be the secondary result of the increase of Ca2+i during ischemia, reflecting reperfusion events set in motion by the calcium transient (Kristiàn and Siesjö 1998). For example, if NO· is produced when the oxygen supply is re-established, the simultaneous presence of NO· and reactive oxygen species, notably ·O2−, can create very harmful chemical species, such as hydroxyl radicals (·OH) and peroxynitrite (Beckman et al., 1990, Samdani et al., 1997).
In this paradigm of secondary damage, triggered by a nonphysiologic Ca2+ transient, the mitochondria occupy a central role. This is not only because mitochondrial dysfunction underlies the bioenergetic failure that triggers the initial loss of cell calcium homeostasis, but also because the mitochondria absorb a major fraction of the cell calcium load once they become re-energized during recovery from an insult, or during reperfusion (Werth and Thayer 1994; White and Reynolds 1995; Schinder et al., 1996). When doing so, they give a spurt of production of reactive oxygen species (Dykens 1994; Dugan et al 1995; Reynolds and Hastings 1995; Piantadosi and Zhang 1996). Furthermore, it is now widely assumed that metabolically strained mitochondria can release molecules that trigger a cascade of reactions leading to cell death, notably apoptosis.
In light of the re-emphasis of the pathogenetic role of mitochondria in brain disease, the article by Paschen and Doutheil (1999) seems almost heretical because the authors put the ER, and the ER calcium homeostasis, into the focus of interest. Supported by extensive literature data, and by their own results, they argue, in a thoughtful and scientifically stringent manner, that the support of the mitochondrial origin of cell death is less than conclusive, and that much speaks in favor of an alternative, ER hypothesis. When addressing this apparent divergence of opinions, in an area of clear relevance to human disease, we will briefly review the mitochondrial hypothesis of (apoptotic) cell death, before discussing the evidence supporting the role of the ER. However, it seems justified to begin by recalling some basic facts about cell calcium metabolism.
CELL CALCIUM REGULATION
Fig. 1 gives a schematic overview of factors regulating calcium homeostasis in internal stores, i.e. ER, calciosomes, and mitochondria. Cell calcium entry by voltage-sensitive and agonist-operated plasma membrane channels leads to an increase in Ca2+i. Such an entry also leads to an increase in the total cell calcium content. Ca2+i also increases if the appropriate agonists (e.g. glutamate) activate surface receptors coupled to phospolipase C and when inositol trisphosphate (IP3), formed during degradation of phosphatidyl inositol bisphosphate, activates IP3 receptors on the ER membranes. This is because IP3 receptor activation leads to release of Ca2+. The ensuing increase in Ca2+i (or, indeed, any increase in Ca2+i) can trigger release of additional calcium from ryanodine- and caffeine-sensitive stores in calciosomes, i.e. non-ER organelles that are insensitive to IP3. Additional intracellular calcium stores have been discussed, but these are neither sensitive to IP3 nor to ryanodine or caffeine, and their calcium content can only be discharged by the calcium ionophore A 23187 (Paschen and Dutheil 1999).
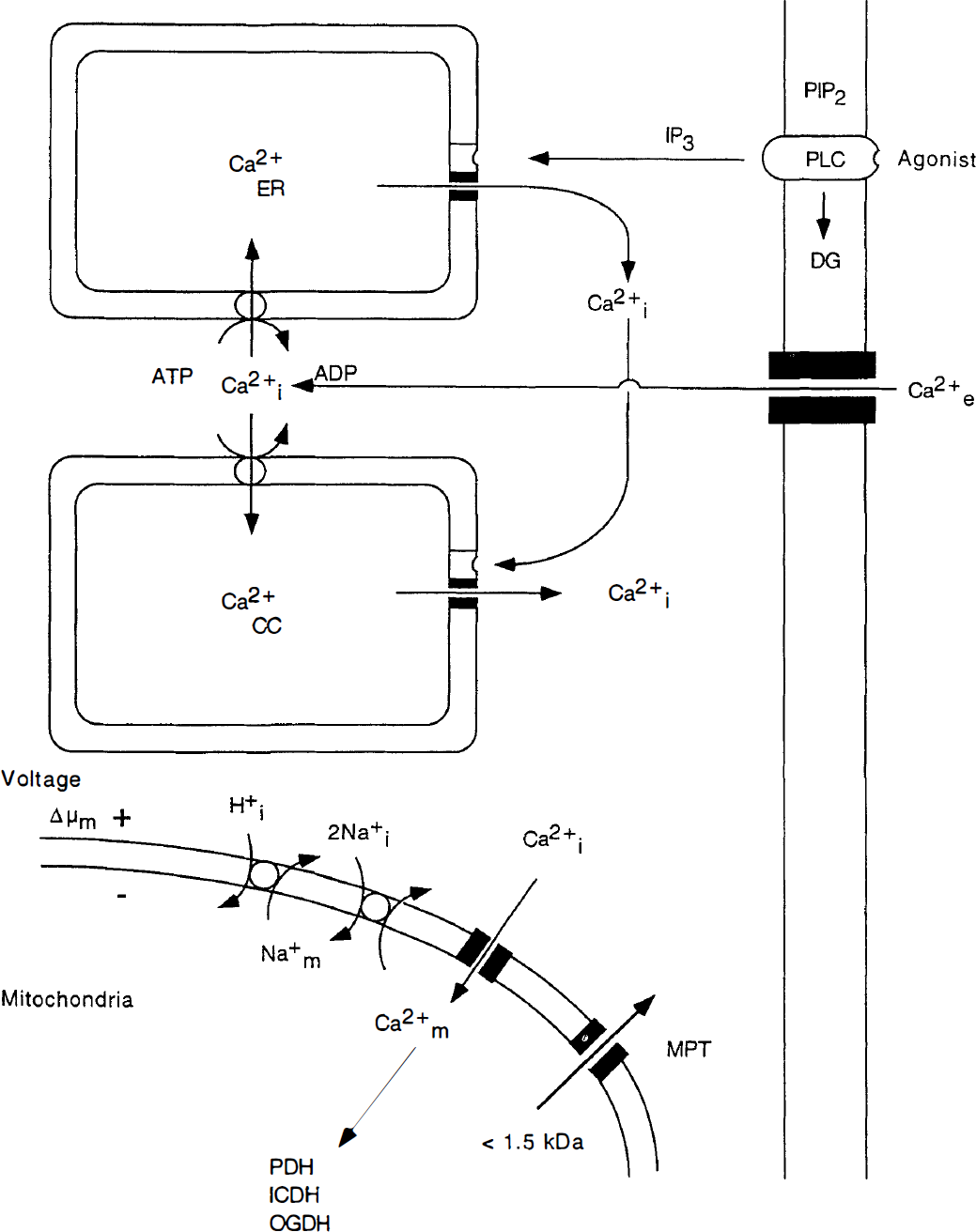
Schematic diagram illustrating calcium fluxes associated with the endoplasmic reticulum (ER) and calciosomes (CC), as well as with mitochondria. The upper part of the figure shows that agonists (e.g. glutamate) acting on surface receptors coupled to phospholipase C (PLC) trigger the breakdown of phosphatidyl inositol bisphosphate (PIP2) to inositol triphosphate (IP3) and diacylglyceride (DG). Inositol trisphosphate activates surface receptors on ER membranes, triggering Ca2+ release. The ensuing increase in Ca2+i can then activate ryanodine receptors on calciosomes, leading to further release of calcium. The ER and the calciosomes are reloaded with calcium by the sarcoplasmic-endoplasmic reticulum Ca2+ – ATPase. These pumps are connected to plasma membrane channels, yielding “capacitative” Ca2+ recharge of the internal stores. The lower part of the figure shows that Ca2+ uptake by the mitochondria occurs by an electrophoretic uniporter, driven by Δμm, while efflux occurs by Ca2+/Na+ exchange, with H+/Na+ exchange restoring the Na+ gradient. Under certain circumstances, a mitochondrial permeability transition (MPT) pore is assembled. This pore, which is blocked by cyclosporin A (CsA) would discharge any Ca2+ accumulated in the mitochondria.
Internal release of calcium causes an increase in Ca2+i but the only event increasing the total cell calcium content is influx through plasma membranes. However, Ca2+ efflux from ER and calciosomes triggers a compensatory influx of calcium by ATP-driven transport that is somehow coupled to a corresponding calcium translocation across the plasma membrane. The transporter, i.e. the sarcoplasmic-endoplasmic reticulum Ca2+-ATPase, is inhibited by the plant alkaloid thapsigargin (T9).
At steady state, Ca2+i is determined by the balance between influx-efflux across the plasma membrane. In the somewhat shorter perspective, the balance between efflux and influx of calcium across the membranes of the ER and other internal stores plays a role in regulating Ca2+i. In an even shorter perspective, the mitochondria (and intracellular calcium buffering) plays an important role in dampening physiologic or pathologic transients.
As shown in the lower part of Fig. 1, calcium is cycled across the inner mitochondrial membrane. Calcium cycling across the membrane normally serves the purpose of regulating Ca2+-dependent intramitochondrial dehydrogenases that control flux in the citric acid cycle, and of buffering large calcium loads during intense activation (Denton and McCormack, 1990; Gunter and Pfeiffer 1990; Nicholls, 1985; White and Reynolds 1995; Wang and Thayer 1996). Ca2+ uptake by the mitochondria occurs by a uniporter, driven by the mitochondrial membrane potential (Δμm). The high capacity uniporter transports calcium at a rate that is proportional to Ca2+i; thus, if the latter increases to micromolar concentrations, large amounts of Ca2+ can accumulate in the mitochondria.
Efflux of Ca2+ from the mitochondria occurs by Ca2+/2Na+ exchange, with coupled Na+/H+ exchange normalizing the Na+ gradient. This is a low-capacity system, suggesting that release of large calcium loads takes time. However, under certain circumstances, a mitochondrial permeability transition (MPT) pore is opened (“assembled”), which is indiscriminately permeable to solutes with a molecular mass of < 1500 d (Gunter and Pfeiffer 1990; Gunter et al., 1994; Zoratti and Szabo 1995; Bernardi and Petronilli 1996). One result of the MPT pore opening is that any mitochondrial calcium load will be rapidly discharged.
MITOCHONDRIA AS TRIGGERS OF CELL DEATH
It is debated whether the MPT has the physiologic function of releasing large mitochondrial calcium loads. What is known, though, is that an MPT pore assembly can lead to mitochondrial dysfunction and cell death (Zoratti and Szabo 1995; Schinder et al., 1996; Zamzami et al., 1996, 1997; Green and Reed 1998; Kroemer et al., 1998; Mignotte and Vayssiere 1998). The basic events during an MPT pore opening encompass equilibration of the electrochemical potential for H+ (ΔμH+) thereby leading to a collapse of Δμm and to cessation of mitochondrial ATP formation (Fig. 2). In fact, dissipation of ΔμH+ leads to a reversal of the F0-F1-ATPase reaction, ATP being wasted in extruding H+. Mitochondrial permeability transition pore opening is inhibited by low intramitochondrial pH, and by the immunosuppressant cyclosporin A (CsA).
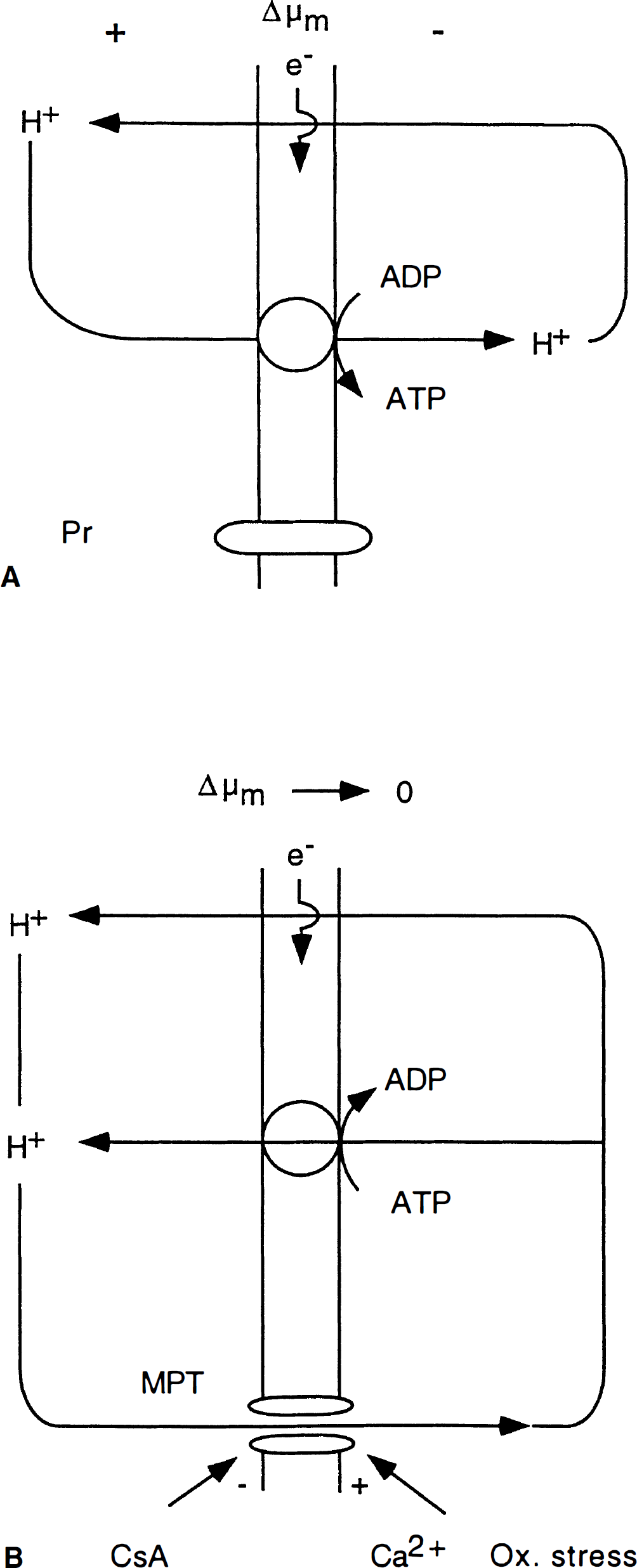
Schematic diagram illustrating H+ fluxes across the inner mitochondrial membrane under normal and pathological conditions.
The adverse conditions leading to an MPT pore opening encompass mitochondrial calcium accumulation and oxidative stress. In view of this, one intuitively feels that ischemia with recirculation sets the stage for MPT-related pathology (Crompton and Andreeva 1993; Duchen et al., 1993; Griffiths and Halestrap 1993; Richter 1993). This assumption received strong support from the results of Uchino et al., (1995, 1998), who showed that CsA, if allowed to pass the blood-brain barrier, virtually eliminated the hippocampal CA1 damage resulting from 7 or 10 minutes of forebrain ischemia in the rat. The results were further supported by those showing that CsA ameliorated the exaggerated ischemic brain damage incurred by hyperglycemic animals, and suppressed the postischemic seizures observed in these animals (Li et al., 1997). FK506, another immunosuppressant that lacks the ability of CsA to block the MPT pore assembly, had a less pronounced effect on CA1 damage after forebrain ischemia (Drake et al., 1996). This suggests that the blocking of an MPT has neuroprotective effects over and above that caused by the immunosuppression.
We have already referred to the fact that the MPT, and perhaps mitochondrial dysfunction in general, constitute important triggers of cell death (Hirsch et al., 1997; Zamzami et al., 1997; Green and Reed 1998; Kroemer et al., 1998). The initial discussion of these events was centered on programmed cell death (apoptosis). However, it is now recognized that the triggers of cell death may be common to apoptosis and necrosis. Thus, a variety of inducers can, dependent on the circumstances, trigger either the classical sequence of events leading to apoptosis or the equally established program causing necrotic cell death (Hirsch et al., 1997; Green and Reed 1998; Kroemer et al., 1998). This concept is applicable also to cells of neuronal origin. In fact, some of the best evidence in support was derived from studies on neurons in primary culture, subjected to transient glutamate exposures (Ankarcrona et al., 1995; Bonfoco et al., 1995). It turned out that brief exposures to moderate glutamate concentrations preferentially caused apoptotic cell death, whereas necrosis dominated after more dense and long-lasting insults. Furthermore, the commitment to either apoptosis or necrosis correlated to the degree of mitochondrial dysfunction.
In discussing cellular events leading to mitochondria-induced cell death, whether of the necrotic or apoptotic type, the following facts should be kept in mind (MacManus and Linnik, 1997; Wyllie, 1997; Green and Reed 1998; Thornberry and Lazebnik 1998).
Much information on the cell death pathway has been obtained on the nematode cenorhabitis elegans in which death and survival genes have been identified. The genes CED-3 and CED-4 promote apoptosis, whereas CED-9 inhibits apoptosis. CED-3 is a caspase, normally existing as a zymogen that can be activated by proteolytic self-cleavage. CED-4 binds to CED-3, promoting CED-3 activation. However, by binding to CED-4, CED-9 prevents it from activating CED-3.
The caspases comprise of a family of cysteine proteases which includes interleukin-1β-converting enzyme (ICE, caspase 1), a protease involved in inflammation. However, the mammalian homologue to CED-3 is caspase-3, the executioner of cell death. The homologue to CED-4 is Apaf-1, and the homologues to CED-9 are anti-apoptotic members of the BCL-2 family (Bcl-2, Bcl-xL). This family also contains proapoptotic members, such as BAX and BIC (Chao et al., 1995; White et al., 1998).
Many of the proteins of the BCL-2 family are bound to the outer mitochondrial membrane. However, BCL-2 is also found on the cytoplasmic face of the plasma membrane, endoplasmic reticulum, and nuclear envelope. This suggests that BCL-2 may “sense” the state and integrity of these membranes, possibly modulating their function by influencing the transmembrane flux of ions, and the release of protein molecules. Furthermore, since BAX is a cytosolic protein, it could influence any membrane facing the cytosol.
Fig. 3, modified from Green and Reed (1998, see also Zamzami et al., 1997; Kroemer et al., 1998) illustrates the cascade of mitochondria-triggered events that either leads to necrosis or to apoptosis. Cell death triggers, such as mitochondrial calcium overload or oxidative stress, can lead to mitochondrial depolarization and an MPT that, in turn, causes the mitochondria to swell. The depolarization and the swelling cause release of mitochondrial proteins, including apoptosis-inducing factor and cytochrome c. If the decrease in Δμm is sustained, the mitochondria remain depolarized and uncoupled, large amounts of reactive oxygen species are formed, and ATP production ceases. The end result is necrosis.
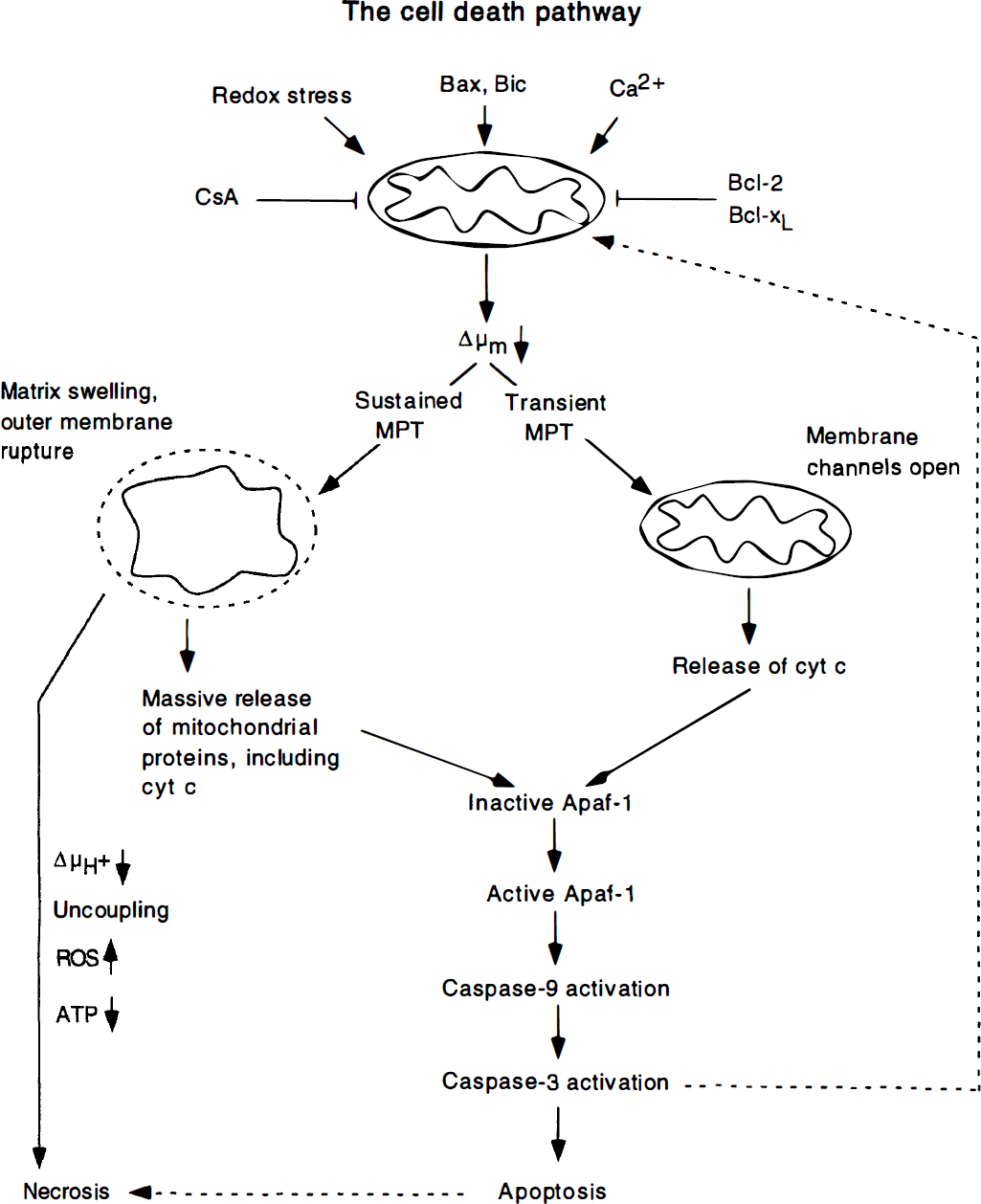
This schematic diagram, modified from Green and Reed (1998), illustrates how mitochondria can trigger necrotic or apoptotic cell death. We envisage that the “cell death trigger” is a decrease in Δμm which is promoted by mitochondrial calcium accumulation or oxidative stress and counteracted by cyclosporin A (CsA) and the anti-apoptotic members of the BCL-2 gene family. A sustained mitochondrial permeability transition (MPT) is apt to cause large amplitude mitochondrial swelling with outer membrane rupture, leading to massive release of mitochondrial proteins (and glutathione). The immediate result is collapse of the ΔμH+, uncoupling of phosphorylation, a spurt of production of reactive oxygen species (ROS) and cessation of ATP production, while the ultimate result is cell necrosis. The right-lower part of the figure shows that apoptotic cell death, spurted by release of cytochrome c and other apoptogenic factors, trigger cell death by a series of events which involve Apaf-1 and activation of caspases, including caspase-3 the “executioner” of cell death.
In a different setting, the mitochondrial depolarization is either reversible or cannot be observed; yet, cytochrome c or other caspase inducers are released, possibly because the death triggers allow channels in the outer mitochondrial membrane to be opened for such release, e.g. by proapoptotic members of the BCL-2 family such as BAX. An MPT may or may not be involved; if it is, one can envisage a transient opening of the MPT pore. However, because the mitochondria maintain their compact structure, the commitment to (apoptotic) cell death is the release of cytochrome c, apoptosis-inducing factor, or mitochondrial procaspases. Such proteins, when released, activate extramitochondrial caspases by binding to Apaf-1 thereby forcing it to associate with procaspase-9, causing caspase-9 to be activated. Once this occurs, caspase-9 can cause other caspases (e.g. caspase-3) to be activated, leading to cell death.
This sequence of events is well established, but the participation of an MPT is largely unclarified. For example, in some systems, caspase activation is known to feed back to the mitochondria, causing them to depolarize. This means that mitochondrial failure may be the result of caspase activation and not its cause.
The sequence of events depicted in Fig. 3 demand additional explanation. We will recall some facts pertaining to the initiation of the death signal and the activation of caspases which, as stated, are the executioners of cell death.
The death signal, committing cells to apoptotic death, can be cell injury or events occurring at surface receptors. Injury to the cell may affect the mitochondria, as in disease conditions causing bioenergetic compromise, but in other instances, the primarily affected structure may be the nucleus (e.g. radiation damage), or the plasma membrane (certain toxins). The death receptors belong to the TNF receptor gene family, the best-known being CD 95 (or Fas or Apo-1). It is not immediately obvious that, in all these conditions, mitochondrial dysfunction is what transmits the death signal to the executioners.
As Fig. 3 shows, release of cytochrome c from mitochondria initiates cell death. Such release seems to provoke the assembly of an “apoptosome”, which is composed of cytochrome c, Apaf-1, and procaspase 9. The result is activation of caspase 9 which then triggers activation of other caspases, including caspase-3. Thus, once cytochrome c is released, the cell is committed to die by a rapid series of apoptotic events involving Apaf-1-mediated caspase activation (Kluck et al., 1997; Nicholson and Thornberry 1997; Reed 1997). However, if large amounts of cytochrome c are released, the result may be necrotic cell death.
The release of cytochrome c is inhibited by the presence of BCL-2, possibly because BCL-2 (and Bcl-xL) can lessen the decrease in Δμm and prevent the MPT. By the same token, the pro-apoptotic Bax may induce apoptosis by enhancing the assembly of an MPT pore. However, an alternative possibility is that antiapoptotic proteins (BCL-2 or Bcl-xL), docking at membrane sites, bind cofactors such as Apaf-1, thereby preventing them from activating procaspase-9, and that proapoptotic members of the family (such as BAX or BIK) act by displacing the cofactors from their binding sites (Adams and Cory 1998).
The importance of the events illustrated in Fig. 3 is shown by experiments in brain ischemia. As already remarked, CsA ameliorates brain damage caused by transient forebrain and focal ischemia, suggesting a beneficial effect of the immunosuppressant on MPT pore opening (Uchino et al., 1995; Li et al., 1997; Butcher et al., 1997). Furthermore, release of cytochrome c has been documented (Ouyang et al., 1998), as has activation of caspase-3 (Hara et al., 1997; Chen et al., 1998). Finally, caspase antagonists (e.g. z-VAD-fmk) have been shown to ameliorate damage caused by forebrain (Chen et al., 1998) and focal (Endres et al., 1997; Hara et al., 1997) ischemia.
In summary, a variety of death triggers initiate a series of events that lead to apoptotic or necrotic cell death. The end result of the cascade of events thus triggered is activation of caspase-3, the executioner of cell death. Although caspase precursor are constitutively expressed in all living cells, inhibitors normally keep the affected proteases inactive, and it takes a pro-apoptotic signal to activate initiator caspases, these in turn activating the effector caspases. The delicate balance between cell survival and cell death is significantly influenced by the pro- and anti-apoptotic influences exerted by members of the BCL-2 gene family. Extensive data suggest that, in upsetting the normal balance, the mitochondria and the MPT play a pivotal role.
THE ENDOPLASMIC RETICULUM AND CELL DEATH
The compelling arguments put together by Paschen and Dutheil (1999) can be examined in light of the information contained in Figs. 1, 2 and 3. Two important facts should be recalled. First, release of Ca2+ from the ER compartments occurs in response to IP3 (compartment 1) and to Ca2+ (compartment 2). Second, release from the first compartment is enhanced by IP3 agonists and blocked by IP3 antagonists, while release from compartment 2, which houses the machinery for folding and processing of proteins, can be blocked by dantrolene.
Evidence in support of the endoplasmic reticulum hypothesis
As reviewed by Paschen and Dutheil (1999) the following facts can be quoted in support of the ER hypothesis of cell death.
In vivo results such as those obtained after transient ischemia show that, in all brain areas affected by the insult, overall protein synthesis rate is depressed. In some ischemia-resistant areas, this depression is transient, but in others, notably the vulnerable ones, it is sustained. At first sight, this could result from the influx-release of Ca2+, increasing Ca2+i to intolerable levels. However, Paschen and Dutheil (1999) argue that this is not the problem, particularly since, at least in vitro, protein synthesis is depressed even if Ca2+ is prevented from increasing by Ca2+ chelators. This leads to the conclusion that it is the decrease in Ca2+ER that leads to inhibition of protein synthesis.
In spite of these results, it can be discussed if the depression of protein synthesis is the cause of the ultimate cell death, or if it is an epiphenomenon. Paschen and Dutheil (1999) submit that the latter may be the case, and argue that the cause of cell death is a dysfunction of ER membranes, reflecting an imbalance between passive efflux of calcium and energy-dependent reentry. Also, ER dilatation is observed in ischemia, obviously preceding necrotic cell death.
The authors dismiss the mitochondrial hypothesis on several grounds. The first one is that CsA, the virtually specific blocker of the MPT pore discussed above, also affects cell ER metabolism and function. For example, CsA is known to induce expression of the ER-resident stress protein grp78, which is an established neuroprotectant. Furthermore, CsA was reported to block TG-induced apoptosis in a setting where ATP concentrations were normal. Finally, additional results suggest the CsA directly modulates the ER calcium pump activity in extracerebral systems in vitro. As a final argument, it was emphasized that, in vivo, both CsA and the immunosuppressant FK506 have proven anti-ischemic effects, although FK506 does not block the MPT pore.
Additional arguments derive from the fact that, in vitro, TG, an irreversible blocker of the ER Ca2+-ATPase, can induce a block of cell division, growth arrest of cells, and apoptosis. TG-induced apoptosis is accompanied by activation of caspase-3 and suppressed by caspase inhibitors such as Z-VAD-fmk. Furthermore, dantrolene, which blocks Ca2+ release through the ER ryanodine receptor channel, is neuroprotective in vivo.
Another argument is that BCL-2 and its protein products are found also in ER membranes. Results have shown that they are able to suppress the depletion of ER calcium stores in several pathologic states, suggesting that this anti-apoptotic gene and its protein products modulate Ca2+ flux between the cytosol and the ER, or between different ER compartments.
The final arguments are based on in vivo experiments on animals subjected to ischemic transients. In these, antagonists of metabotropic glutamate receptors proved to be neuroprotective, whereas IP-3 receptor agonists aggravate tissue damage. It is also of interest that IP3 receptor agonists, when injected intracerebroventricularly, induce seizures and brain damage.
Reservations
In spite of these compelling arguments it cannot be accepted without reservations that the ER is a primary trigger of cell death. We wish to raise the following points.
Admittedly, the immunosuppressant CsA does not act exclusively on mitochondrial membranes. Like FK506, it suppresses calcineurin, a phosphatase involved in NO metabolism; furthermore, both immunosuppressants antagonize rotamase activity, i.e. they act to suppress leucine cis-trans isomerase activity, required for protein folding (e.g. Steiner et al., 1997). What is unique to CsA is that it blocks the MPT pore. FK506 does not do that. However, it has been reported that FK506 blocks an unspecific large conductance channel in ER membranes. This could explain why both CsA and FK506 “stabilize” membranes whose integrity is essential for cell survival. However, it does not identify the ER as a trigger of apoptotic cell death.
As stated, BCL-2 is not exclusively localized to mitochondrial membranes but is also found on the cytosolic side of ER plasma and nuclear membranes. This suggests that anti-apoptotic members of the BCL-2 family act to stabilize membranes, e.g. by protecting them from free radical attack (Kane et al., 1993), and to prevent release of apoptogenic factors to the cytosol and activation of cytosolic caspases. Again, this does not prove that ER membrane dysfunction is a leading event in the triggering of apoptosis, to the exclusion of mitochondria. Thus, although the apoptosis-inducing effect of TG is suggestive, it has not been established that ER dysfunction, and the signals this may create, precedes mitochondrial dysfunction and release of mitochondria-derived apoptogenic factors. At best, the results suggest that ER dysfunction contributes to cell death.
Compelling evidence suggesting a mitochondrial origin of necrotic and apoptotic cell death is that necrosis seems to require failure of mitochondrial ATP production and apoptotic release of mitochondrial factors, such as cytochrome c, apoptosis-inducing factor, and procaspases. That these are of mitochondrial origin is shown by the fact that mitochondria, when subjected to depolarizing conditions, can induce all the morphologic signs of apoptosis in isolated nuclei. Conceivably, similar factors can be released from metabolically perturbed ER membranes, but this has not yet been shown.
Other evidence, equally compelling, show that when neurons in vitro are subjected to glutamate transients, they are protected by drugs that inhibit calcium uptake by the mitochondria (Stout, 1998). Because these drugs do not inhibit calcium influx into cells, or the increase in Ca2+i levels, the results suggest that mitochondrial calcium uptake is of decisive pathogenetic importance.
In summary, the evidence discussed by Paschen and Dutheil (1999) question the overriding importance of mitochondria in triggering apoptosis-necrosis and puts the ER calcium metabolism, and the ER membranes, into the focus. Clearly, they can lean on the facts that TG, which has no known effects on the mitochondria, can induce apoptosis, and that BCL-2 resides on the cytoplasmic face of ER membranes, obviously influencing ER membrane function. Further support for their hypothesis is that ischemic cell death is ameliorated by IP3 antagonists and by dantrolene, a known blocker of ryanodine receptors. However, although these results suggest that ER dysfunction contributes to brain damage after ischemic transient, they do not invalidate the mitochondrial hypothesis of necrotic-apoptotic cell death. It seems highly justified that additional information is obtained on this subject, particularly on any proapoptotic factors released from the ER.