
Abstract
We report the case of a 78-year-old patient with late diagnosis of hyperoxaluria type III (PH3). He developed renal failure after nephrectomy for clear cell papillary renal carcinoma and complained of recurrent urolithiasis for some 30 years, whose aetiology was never identified. Biochemical laboratory investigations of urine and urolithiasis composition revealed marked hyperoxaluria but normal concentrations of urinary glyceric and glycolic acid as well as stones of idiopathic calcium-oxalate appearance. Furthermore, the dietary survey showed excessive consumption of food supplements containing massive amounts of oxalate precursors. However, the persistence of excessive hyperoxaluria after his eating habits was changed leading us to perform molecular genetic testing. We found heterozygous mutations of the recently PH3-associated HOGA1 gene when sequencing PH genes. This is the first description of late diagnosis primary PH3 in a patient with several additional pro-lithogenic factors. This case illustrates the importance of undertaking a complete biological work-up to determine the aetiology of hyperoxaluria. This may reveal underdiagnosed primary hyperoxaluria, even in older patients.
Introduction
Hyperoxaluria is a major risk factor in calcium-oxalate nephrolithiasis, which occurs in about 10% of the population. Primary hyperoxaluria (PH) is a group of underdiagnosed autosomal-recessive inherited disorders characterized by increased synthesis and excretion of the end-product oxalate.1,2 High urinary oxalate excretion results in poorly soluble calcium-oxalate crystallization within the urinary tract, the renal parenchyma (nephrocalcinosis) and/or recurrent urolithiasis, the clinical hallmarks of PH. PH1 (MIM# 259900) is the most frequent and most severe clinical form of PH owing to enzyme alanine glyoxylate amino-transferase (MIM 604285) deficiency, a peroxisomal enzyme involved in glyoxylate detoxification. PH2 (MIM# 260000) is associated with enzyme glyoxylate reductase/hydroxypyruvate reductase (MIM 604296) deficiency and has a less severe course. Recently, PH3 (MIM# 613616) was reported as being due to mutations in the 4-hydroxy-2-oxoglutarate aldolase gene (HOGA1, formerly known as DHDPSL; MIM# 613597). 3 PH is usually diagnosed early in childhood as recurrent urolithiasis and nephrocalcinosis, and the diagnosis is usually established on biochemical findings, urolithiasis structural analysis and is confirmed by molecular genetic studies. We report here the first case, to our knowledge, of late-onset diagnosis of PH3 in a 78-year-old patient.
Case presentation
A renal clear cell papillary carcinoma affecting the right kidney was fortuitously discovered by ultrasound imaging of the urinary tract in a 78-year-old man. A nephrectomy was performed, but his renal function deteriorated rapidly during postoperative recovery (basal serum creatinine increased from 137 µM to 223 µM, estimated glomerular filtration rate (eGFR) 18 ml/min/1.73 m2). Concerning his medical history, he had chronic kidney disease of unknown origin, arthrosis, spine surgery, atrial fibrillation and arterial hypertension. He also had suffered from urolithiasis for over 30 years that required ureterorenoscopy and extracorporeal lithotripsy. The stone disease was never investigated and aetiology remained undetermined. He was referred to the nephrologist for kidney management and biological investigations.
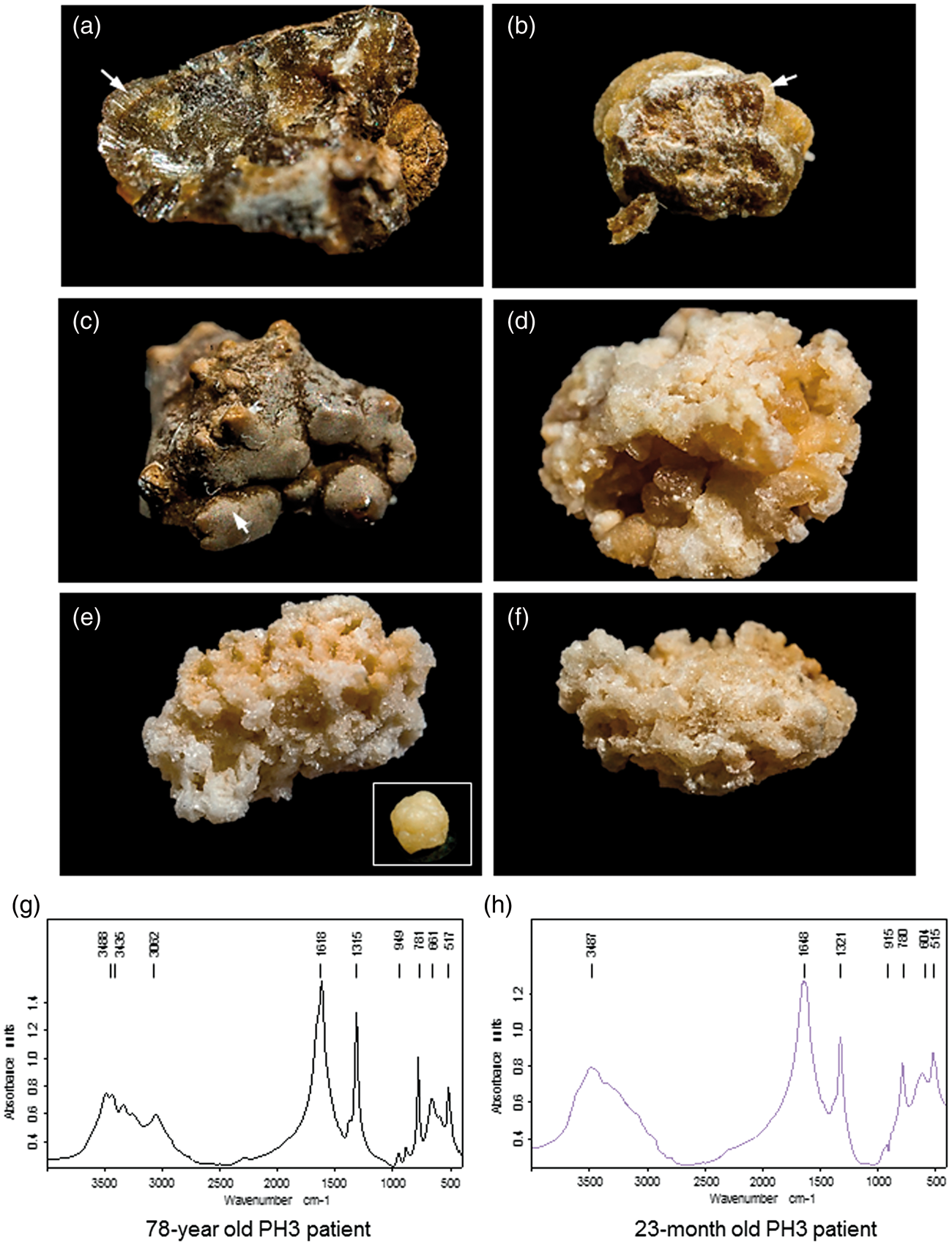
In this patient, serum analysis revealed elevated creatinine and urea without an abnormal phospho-calcic metabolism or electrolyte pattern (Figure 1(a)). However, several pro-lithogenic conditions were identified in his urine such as hypocitraturia (0.6 mmol/24 h; n = 2.2–2.4 mmol/24 h), hypomagnesuria (2.1 mmol/24 h; n = 3–6 mmol/24 h) and nocturnal hydropenia and especially a significant hyperoxaluria (1093 µmol/24 h; urinary oxalate to creatinine ratio = 102 µmol/mmol; n < 40 µmol/mmol), which was associated with calcium-oxalate monohydrate (COM, Whewellite) crystalluria. We examined both the composition and morphological characteristics of his stones by using stereomicroscopy and infrared spectroscopy (Figure 2(a) to (c)). The stones had a dark-brown surface and well-organized radiating inner structure, which are the morphological features of typical idiopathic common whewellite stone. However, superficial grey crystalline aggregates and pale-yellow radiating structures in some of them suggested active lithogenesis and major episodes of hyperoxaluria. Infrared spectroscopy revealed a pure COM composition (>98%) (Figure 2(g)).
(a) Plasmatic and urinary parameters from 24 h urine samples and (b) schematic representation of (⋄) serum creatinine (µmol/L) and (○) urinary oxalate (µmol/24 h) concentrations over the course of the disease (weeks after initial presentation). Crystalluria was analysed on first morning urine by polarized light microscopy. Urinary oxalate and citrate concentrations were determined using ion-exchange HPLC with conductivity detection. Urinary glycolate and glycerate were analysed by a GC-MS method. Other biochemical parameters were determined using standard clinical laboratory methods. (a, b, c) stones from the PH3 case report exhibited well-organized surface areas of dark-brown colour and a radiating structure. Arrows indicate (c) grey crystal aggregates and (b) light colour areas on stone surface that would suggest active lithogenesis. (g) Infrared spectroscopy analysis indicated a pure calcium-oxalate monohydrate composition. Stones from infantile PH1 (d) and infantile PH3 (e, f) have a light surface colour are inhomogeneous and have crystal aggregates of various size. (e, f, h) Infantile PH3 stones exhibit fine-grained calcium-oxalate monohydrate regions and large crystalline calcium-oxalate dehydrated bipyramidal regions, as confirmed by infrared spectroscopic analysis.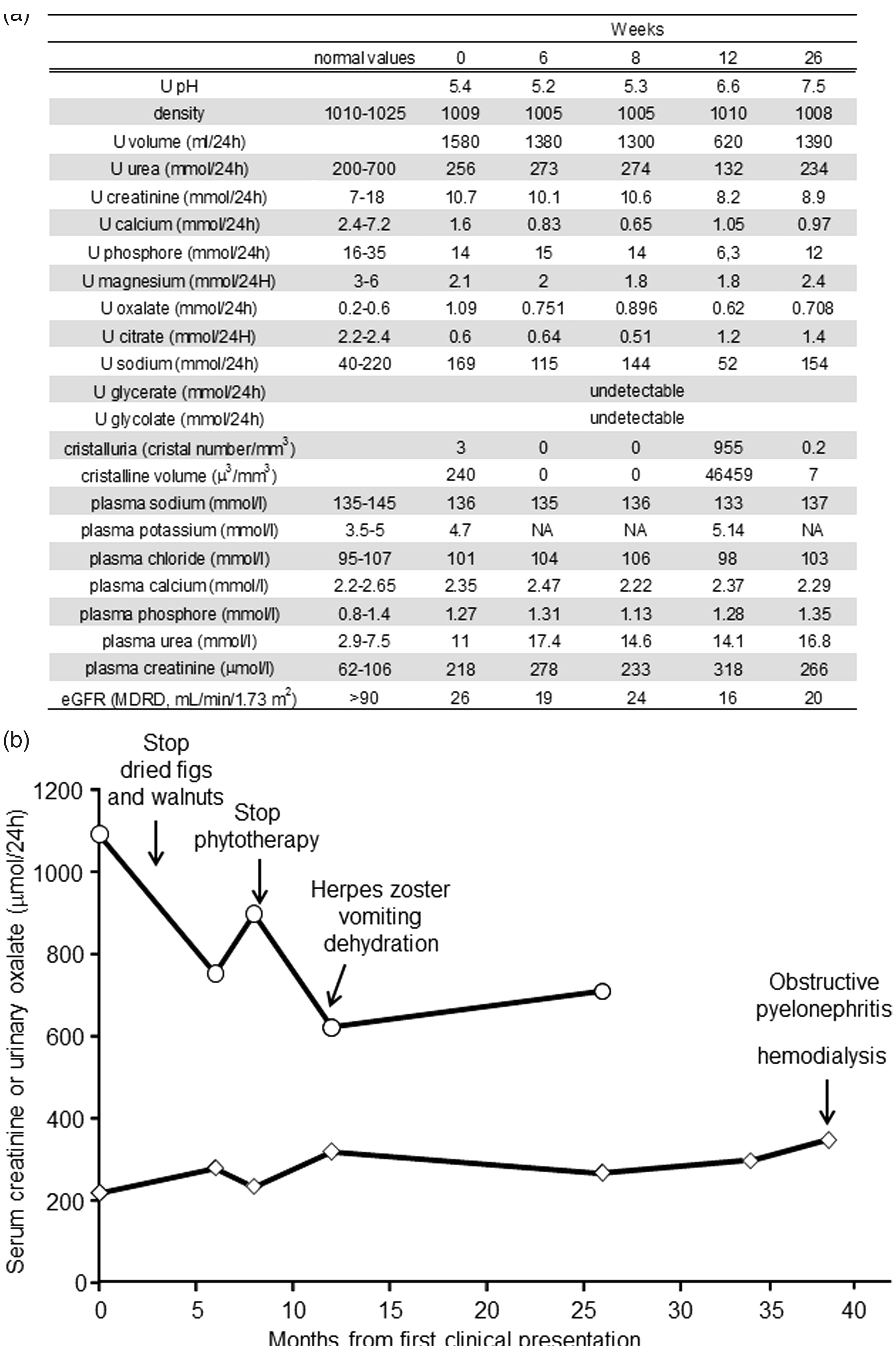
Dietary oxalate and the metabolism of hydroxyproline, glycine and glyoxal contribute to the urinary excretion of oxalate.4,5 An investigation of his diet revealed peculiar eating habits with the daily consumption of oxalate-containing food such as walnuts and dried figs throughout the evening. Furthermore, he had regularly consumed phytotherapy supplements for arthrosis and prostate hyperplasia-related dysuria for several months. Such products contain meadowsweet (Filipendula ulmaria), a medicinal plant providing ascorbate, which is subsequently broken down into oxalate. 6 Another phytotherapy product he was taking contained shark cartilage, a significant source of hydroxyproline, which is a precursor of oxalate metabolism.4,5 He was given advice how to reduce his dietary intake of oxalate, and crystalluria analyses were then performed regularly for three years to assess the efficacy of the dietary advice and avoid any recurrence of stones (Figure 1(b)). While a significant decrease in oxalate excretion was observed, his oxaluria remained elevated (>600 µmol/24 h or >70 µmol/mmol creatinine, normal <40 µmol/mmol). No other cause of secondary hyperoxaluria such as enteric hyperoxaluria, vitamin B6 deficiency or massive intake of ascorbate was identified in the patient.
Twelve months after the initial evaluation, he was treated with tramadol for painful herpes zoster and referred to the nephrologist for functional acute renal failure due to intense tramadol-induced vomiting and dehydration. During this episode of decompensating chronic kidney disease, we decided to search for PH. Although urinary glycolate and glycerate excretion are helpful non-specific markers of PH types I and II, respectively, they were undetectable in the patient (Figure 1(a)). According to the frequency of PH1, sequencing of AGXT gene was first performed and revealed no pathogenic mutation. HOGA1 gene was then studied with 5′ and 3′ primers targeting each of the six exons and flanking splice junctions and two heterogeneous mutations were detected: the previously described non-sense mutation c.208C > T mutation (p.Arg70X) and the novel missense substitution c.634A > G (p.Thr212Ala). 7 According to prediction software (AlignGVGD, SIFT, Mutation taster), this second substitution is predicted to affect protein function. In view of these findings, we confirmed the diagnosis of PH3 in this 78-year-old patient and identified both the genetic determinant and precipitating factors responsible for his renal stone disease. He underwent symptomatic treatment based on hyperhydration, sodium bicarbonate and magnesium supplementation. Unfortunately, he developed acute renal failure due to obstructive pyelonephritis related to a stone in the pelvic urethra. Renal function did not recover in spite of adapted antibiotherapy and double-J stent placement, and the patient developed end-stage renal disease (ESRD) for which he is currently receiving hemodialysis.
Discussion
Considering its rarity, PH may go unrecognized for several years after the onset of symptoms. Concerning PH1, the most prevalent and most severe form, there is a high rate of late diagnosis in the setting of advanced renal failure of after kidney graft failure. Most PH3 patients present early onset of the disease (median <2 years), and recurrent urolithiasis and urinary tract infections are the leading symptoms.3,4,8–10 While PH3 usually presents with a milder phenotype than PH1 and PH2 regardless of renal function, eGFR is impaired in some children with PH3. 10 Despite marked hyperoxaluria, only one case of PH3 has been associated with ESRD to date in an early-onset disease patient. 11 To our knowledge, this is the first report of late diagnosis of PH3 in an elderly patient and leading to ESRD. We think that his chronic kidney disease was at least partially due to an oxalic nephropathy. Finding of calcium-oxalate crystals in a kidney biopsy specimen may also suggest PH. 7 Unfortunately, histological sections of the kidney were not analysed to search for calcium-oxalate deposits after the nephrectomy. The HOGA1 enzymatic deficiency was likely to be particularly important regardless to the contribution of eating habits to oxalate excretion in our PH3 patient. HOGA1 is involved in the catabolism of hydroxyproline derived from endogenous and dietary sources and contributes to glyoxylate, glycolate and glycine concentrations in humans.12,13 While the exact pathogenetic mechanism is unknown, HOGA1 deficiency is associated with oxalate overproduction in PH3 patients and excessive hydroxyproline intake may worsen hyperoxaluria. 4 We feel that the function of the remaining kidney rapidly deteriorated after nephrectomy, because the contralateral kidney suffered from several years of chronic kidney disease. Furthermore, massive oxalate excretion and dehydration leading to oliguria may have precipitated deleterious intraparenchymal oxalate deposition and ultimately favoured acute renal failure.
Laboratory input is essential in identifying the aetiology and in establishing the therapeutic monitoring of stone formers. Crystalluria examination is useful for assessing the main metabolic factors involved in crystal and stone formation and allows simple monogenic crystallogenetic pathologies to be identified. It involves the search for: identification and counting of crystals in fresh morning urine as well as biochemical analyses of urine composition and serum. In this patient, we found a positive crystalluria composed of COM crystals which are usually associated with urinary oxalate oversaturation. Hyperoxaluria is the hallmark of PH, but urinary oxalate concentrations retrieved in PH3 overlap those observed in PH1 and PH2. Although non-specific, urinary metabolites such as glycolic acid and glyceric acid may be helpful to discriminate between PH1 and PH2. Urinary glycolic acid is elevated in 2/3 of PH1 patients and may also be elevated in some PH3 patients. 12 Urinary glyceric acid seems to be specifically elevated and pathognomonic in PH2 patients. 14 In our case, neither urinary glycolate or glycerate were elevated. Morpho-constitutional analysis of urinary calculi, i.e. morphologic examination combined with Fourier transform infrared spectroscopy, is decisive for the diagnosis of PH. The majority of PH patients present with urolithiasis and their stones have been reported with peculiar appearance as compared with idiopathic calcium-oxalate stones.15–17 While idiopathic calcium stones are dense, strongly pigmented and well organized with a radiating inner structure, PH stones have a light surface colour, are inhomogeneous and have crystal aggregates of various sizes. While stones from PH1 patients under treatment are compact and composed of COM, stones from untreated PH1 patients contain a wide size range of aggregated calcium-oxalate dihydrate (COD, weddellite) crystals. 15 On the contrary, PH3 stones appear heterogeneous and are composed of aggregated COM and COD, regardless of the treatment. 15 In our experience, we have found similar findings on stones from infantile PH1 and PH3 patients (Figure 2(d) to (f)). Infantile PH3 was previously diagnosed at our medical centre based on recurrent urolithiasis and urinary tract infection, hyperoxaluria, positive crystalluria (COM crystals) and stone analysis in a 23-month-old girl. HOGA1 gene analysis revealed homozygosity for the c.700 + 5G > T mutation and confirmed PH3. Her stones exhibited fine-grained COM regions and large crystalline COD bipyramidal regions (Figure 2(e) to (h)). However, the stones in the present case report had an unusual morphology that did not suggest the diagnosis of PH (Figure 2(a) to (c)). They had well-organized surface areas of dark-brown colour, a radiating structure and were composed of pure whewellite (Figure 2(a) to (c) and (g))). Grey crystal aggregates and light colour areas on a stone surface would suggest active lithogenesis (Figure 2(b) and (c)).
This case illustrates the difficulty of diagnosing PH in patients with recurrent calcium-oxalate urolithiasis. PH is usually suspected based on specific clinical and biological features such as crystalluria, calculi analysis and especially persistent hyperoxaluria. However, the identification of urinary biomarkers would allow biochemical screening for PH3 in subjects with a low index of suspicion, especially among calcium-oxalate stone formers. In this regard, 4-hydroxy-glutamate, an intermediate compound of hydroxyproline metabolism, was recently found to be elevated in PH3 patients due to HOGA1 enzymatic deficiency although a large cohort study would be needed to define its predictive value.18,19
Footnotes
Acknowledgments
We thank Catherine Carlier and Carole Hazera for their expertise and help in analysing stone morphology and composition.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Written consent was obtained prior to using data from the patient’s file and publication, and care has been taken so that no information indicates his identity.
Guarantor
ER.
Contributorship
JH, BL and RDLF were the attending physicians, ER and JMB were medical biologists and performed conventional biochemical and stone analysis, CA performed molecular genetic analysis, SB provided stone pictures. ER researched the literature and wrote the manuscript, RDLF supervised the case study, JH, BL, RDLF, JMB, SB, CA, ER corrected the manuscript. All authors reviewed and approved the final version of the manuscript.