
Abstract
Energetic materials have widespread applications in military, aerospace, and other high-stakes domains. Accurate prediction of their explosive properties is critical for both material development and safe deployment. This paper proposes a Directional-Aware Graph Attention Network (DAGAN) model, which constructs node and edge representations incorporating fine-grained features such as atomic type distributions and chemical bond topological environments. A directional-aware graph attention architecture is designed and integrated with an adaptive training algorithm to enable deep mining of intrinsic molecular characteristics. Experimental results show that the DAGAN model, after hyper-parameter optimization, significantly outperforms traditional machine learning methods such as SVM, RF, and XGBoost in predicting explosive performance. Its attention mechanism effectively captures both local atomic interactions and global structural features, overcoming the limitations of incomplete information in conventional feature engineering. This work offers a novel perspective and method for the research and development of energetic materials.
Keywords
Introduction
Energetic materials play an indispensable role in key areas such as national defense Zeman S and Jungová M, 1 aerospace Wang Y et al., 2 industrial blasting Ahmed and Malik, 3 and pyrotechnics Guo Z et al. 4 Their performance directly affects the efficiency of weapons systems, propulsion in spacecraft, and the safety and the effectiveness of explosive-related applications. The properties of energetic materials are largely determined by their chemical structures, which govern the way energy is stored and released, as well as the material’s sensitivity to external stimuli such as temperature, pressure, impact, or friction Yuan W L et al. 5 Traditionally, the development of energetic materials has relied heavily on extensive experimental testing, which is time-consuming, labor-intensive, and costly. By building reliable computational models to predict material properties, researchers can screen and evaluate a large number of potential candidates before physical experiments, significantly reducing development time and cost. In the search for new energetic materials, computational prediction allows for the early elimination of low-performance candidates, enabling researchers to focus resources on more promising ones. Moreover, accurately predicting molecular properties facilitates a deeper understanding of the structure–property relationships, providing valuable theoretical guidance for performance optimization Elton et al. 6 ; Tian et al. 7 ; Wespiser and Mathieu 8 ; Zang et al. 9
Previous studies have primarily relied on quantum chemical simulations, empirical formulas, or simple structural feature analyses to predict the properties of energetic materials. For example, Mathieu 10 investigated the relationshipsamong detonation velocity, detonation pressure, and impact sensitivity log(H50), finding that log(H50) increases linearly with D-4 and P-2. These predictive models showed good agreement with experimental data for non-aromatic nitro compounds. Bondarchuk 11 analyzed factors influencing the detonation characteristics of C-H-N-O explosives and proposed empirical formulas based on solid-phase enthalpy of formation and crystal density, enabling property estimation using handheld calculators.
However, empirical formulas typically disregard detailed chemical structures and are only applicable to energetic materials with similar structural motifs. They fail to fully capture molecular complexity and the subtle interactions between atoms, limiting the predictive accuracy of such models. Furthermore, methods based solely on elemental composition and basic bond-type statistics often overlook critical information such as atomic spatial arrangements and electronic effects of chemical bonds, which results in substantial errors when predicting complex properties.
With the rise of artificial intelligence, machine learning has emerged as a transformative tool in materials discovery by providing accurate property predictions at significantly reduced computational cost. Chen et al. 12 introduced the concept of spatial matrix descriptors, constructing the Volume Occupancy Matrix and the Heat Contribution Matrix to represent molecular structures. They applied a range of machine learning algorithms—including LASSO Ranstam and Cook, 13 Kernel Ridge Regression Vovk, 14 Bayesian Ridge.
Regression Shi et al., 15 Support Vector Regression Awad et al., 16 Random Forest Regression Rodriguez-Galiano et al., 17 and K-Nearest Neighbors Kramer O 18 to predict energetic material properties. Model performance was evaluated using Leave-One-Out Cross-Validation, which enhanced prediction accuracy, particularly in scenarios with limited data. Zhang et al. 19 employed machine learning techniques to predict thermal decomposition temperatures and investigate their correlation with the thermal stability of energetic materials. Molecular descriptors and Molecular ACCess System (MACCS) fingerprints were generated by RDKit Landrum, 20 and the SHAP (SHapley Additive exPlanations) method was utilized to select 20 key descriptors. Various regression algorithms, including Kernel Ridge Regression (KRR), LASSO, and Random Forest (RF), were applied, demonstrating that the thermal decomposition process is influenced by molecular composition, electronic distribution, chemical bond properties, and the nature of substituents. Davis et al. 21 introduced the MolDensity model, which combines RDKit-generated descriptors with machine learning models trained using Elastic Net Regression Hans, 22 Random Forest Rodriguez-Galiano et al., 17 and Gradient Boosting Trees Ke et al. 23 This model was used to predict critical performance parameters of high explosives, such as crystal density, heat of formation, detonation velocity, and detonation pressure. Liu et al. 24 combined density functional theory calculations with machine learning methods (AdaBoost, SVR, RF, KRR) to accurately predict the impact sensitivity and detonation performances of energetic materials based on 28 physicochemical features, which identified the optimal ranges of key features such as oxygen balance, density, HOMO level, and lipophilicity.
The aforementioned studies typically use molecular fingerprints or simple descriptors as feature inputs for machine learning models. However, these representation methods often suffer from issues such as information loss or the curse of dimensionality. While molecular fingerprints can capture some aspects of molecular structure, they have limited ability to distinguish fine-grained details in complex molecules. Conversely, high-dimensional descriptor vectors may lead to difficulties in model training and increase the risk of overfitting. Moreover, model interpretability remains a significant challenge which deep learning models are often regarded as black boxes, with their internal mechanisms still poorly understood. In contrast, graph neural networks, which represent atoms as nodes and their interactions as edges, offer a more natural and expressive way to study the internal structure and properties of energetic materials.
Hu et al. 25 utilized transfer learning with a Force-Field-inspired Transformer Graph Neural Network (FFiTrNet) to predict the properties of energetic materials. The model was initially trained on a dataset of CHNOF compounds and then fine-tuned on a smaller dataset of enthalpy-related energetic materials. Results showed that transfer learning significantly improved the accuracy of enthalpy predictions—both the Mean Absolute Error (MAE) and Root Mean Square Error (RMSE) were reduced compared to models trained directly on the small dataset. Nguyen et al. 26 compared expert-designed features with molecular representations automatically learned by graph neural networks and found that the Message Passing Neural Network (MPNN) outperformed Random Forest and Partial Least Squares Regression in predicting crystal density. Buterez et al. 27 proposes graph neural network transfer learning strategies based on adaptive readout functions, leveraging low-fidelity data to improve sparse high-fidelity molecular property prediction performance in drug discovery and quantum mechanics tasks with an order of magnitude less high-fidelity data. Yang et al. 28 evaluated three machine learning models—Support Vector Machines (SVM), Random Forest (RF), and Graph Neural Networks (GNNs)—using only molecular topology to predict the density of high-energy compounds. The results demonstrated that GNNs achieved higher accuracy and lower computational costs compared to traditional density functional theory-based quantitative structure–property relationship models. Gao et al. 29 proposed a molecular descriptor-enhanced GNNs model for predicting detonation heat, detonation velocity, and detonation pressure of energetic molecules.By integrating sequence-based molecular descriptors with structure-based graph embeddings, the model captured a more comprehensive representation of molecular features, thereby improving prediction accuracy.
The Directional-Aware Graph Attention Network (DAGAN) model proposed in this work is an original architecture designed by the authors, which constructs node and edge representations incorporating fine-grained features to address the limitations of existing graph neural networks in capturing fine-grained molecular structural features. DAGAN represents molecules as graphs, where atoms are modeled as nodes and chemical bonds as edges. This structure allows the model to naturally capture atomic relationships and reflect the true 2D connectivity and intramolecular interactions. The attention mechanism in DAGAN assigns varying levels of importance to different atoms and bonds, enabling the model to focus on interactions most relevant to material properties. Subtle changes in the local molecular environment can significantly impact the properties of energetic materials, which is of great importance Liu et al. 30 Compared to Graph Convolutional Networks Velickovic et al. 31 ; Zhang et al. 32 ; Chen et al., 33 DAGAN provides greater flexibility in weighted node interactions, effectively capturing subtle differences in molecular behavior and improving prediction accuracy. We will explore how the graph attention mechanism can distinguish key features of energetic materials and compare DAGAN with traditional methods such as RF, 34 SVM, 35 and XGBoost 36 to evaluate the model’s predictive capability and generalization ability across different types of materials.
Experimental section
Data processing
The dataset used in this study is derived from the energetic material molecular structures and their properties reported in the literature by Gao et al.
29
For molecular graph representation in DAGAN, we utilize the RDKit toolkit to convert the SMILES37,38 strings of molecules into 2D conformations. These 2D molecular files are then transformed into graph structures using the Pymatgen
39
library, where each atom is treated as a node and chemical bonds between atoms are treated as edges. Atomic features used as node attributes are obtained via the Mendeleev package and include atomic type, atomic number, functional group, period, formal charge, electronegativity, atomic radius, atomic volume, electron affinity, and first ionization energy. These features are divided into categorical features and continuous features for targeted preprocessing.Categorical features includes atomic type, functional group, period, and hybridization, which are converted into one-hot encoded vectors to avoid artificial ordinal relationships between discrete categories. Continuous features includes atomic number, electronegativity, atomic radius, atomic volume, electron affinity, and first ionization energy. To eliminate the impact of dimensional differences and scale inconsistencies on model training, these continuous features are standardized (Z-score normalization) using the following formula:

Molecular fingerprinting algorithms convert molecules into fixed-length vectors by encoding atoms and chemical bonds within molecular structures. These fingerprints can partially capture structural characteristics of molecules and serve as global feature vectors. In our approach, molecular fingerprints are integrated with the molecular graph representation and concatenated with the final output layer of the DAGAN model. This enables the model to leverage both local and global information prior to making predictions. We use RDKit and DeepChem toolkits to extract molecular fingerprint features, selecting four types: MACCS, Daylight, Extended-Connectivity Fingerprints (ECFP), and Topological Fingerprints (TopoFP). Among them, the MACCS fingerprint represents molecules using a predefined set of structural keys, typically encoded as binary features. Each of the 166 MACCS keys corresponds to a specific chemical substructure. If the substructure is present in a molecule, the associated bit in the fingerprint vector is set to 1; otherwise, it is set to 0.
The Daylight fingerprint is generated by identifying all possible chemical substructures within a molecule. Each substructure corresponds to a specific bit in the fingerprint vector, which is set to 1 whenever the substructure is present.
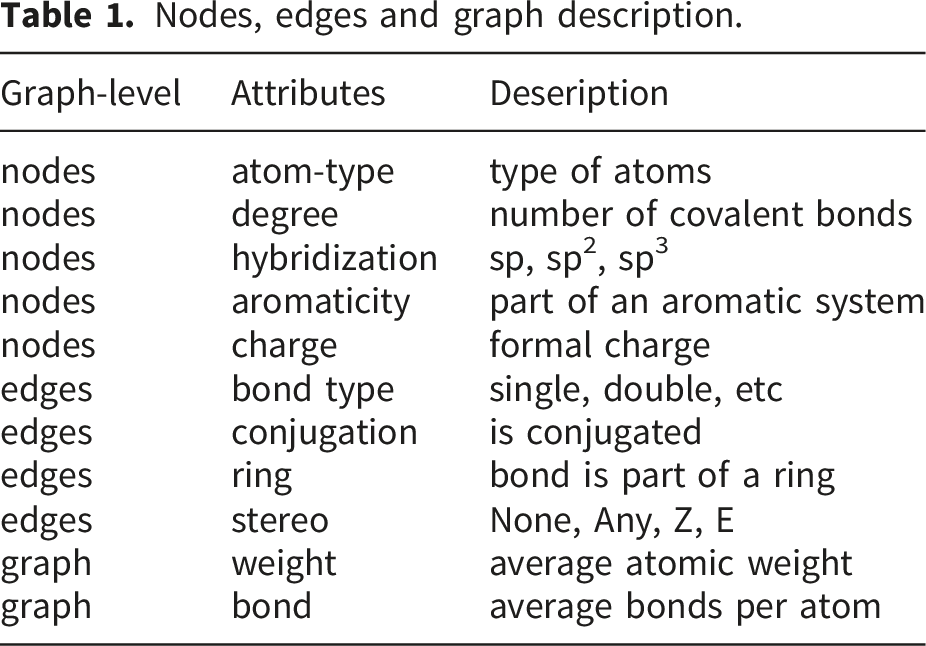
Nodes, edges and graph description.
Dataset splitting
We divide the dataset into training, validation, and test sets based on a predefined ratio: 70% of the data is used for training, 15% for validation to tune hyperparameters, and the remaining 15% for testing the final performance of the model. To minimize bias and ensure the reliability of the results, the dataset is partitioned such that the distributions of molecular structures and properties remain consistent across the three subsets. The three datasets are divided in the same way, and all explosion performance predictions are based on the same datasets. This strategy helps avoid data skew and ensures fair and meaningful evaluation. To evaluate the stability and reliability of the model, all experiments are repeated five times using different random seeds, and the average results are reported.
Evaluation metrics
For the prediction of energetic material properties, we employ several evaluation metrics to comprehensively assess model performance, including MAE, RMSE, and the coefficient of determination R2. These metrics collectively evaluate the closeness of the predicted values to the actual experimental values, as well as the overall accuracy and consistency of the model’s predictions.


GNNs model architecture
Graph Neural Networks (GNNs) adopt a message-passing framework in which atoms are represented as nodes and chemical bonds as edges. Each node u maintains a representation 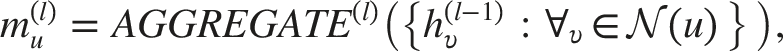

GAT model architecture
Graph Attention Networks (GATs) adopt a message-passing mechanism, where the state of each atom is updated based on information from its neighboring atoms and the associated bond features at each layer, as shown in Figure 1. Specifically, an atom receives messages from its neighbors that include both neighbor atom features and bond features. These messages are aggregated through attention-weighted summation, allowing the model to assign different levels of importance to different neighbors. The aggregated information is then passed through a nonlinear transformation to compute the updated atomic representation. The GAT diagram.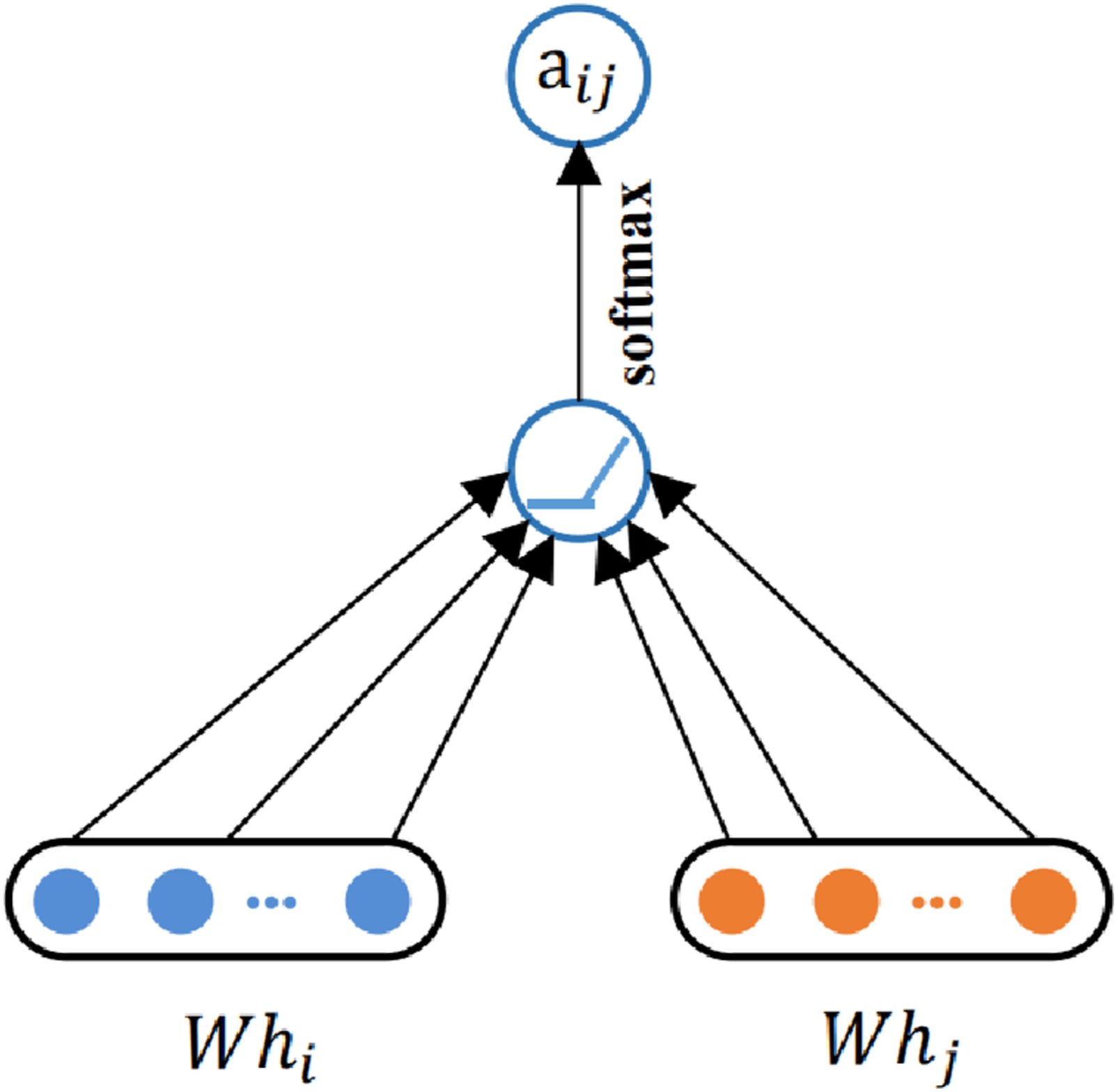
To further enhance the model’s expressive power, the fused features are processed using a multi-layer perceptron, enabling complex interactions to be captured and facilitating deeper feature abstraction. This attention mechanism allows GAT to dynamically focus on the most relevant local interactions, improving its capability to model subtle molecular structures and reactivity patterns.
Bond features are also updated at each layer of the GAT, taking into account the changing states of the connected atoms. Updating bond representations facilitates a more accurate modeling of interatomic interactions, thereby enhancing the model’s ability to understand molecular structures. GAT achieves this by stacking multiple graph attention layers, where at each layer, attention coefficients are computed for each atomic pair 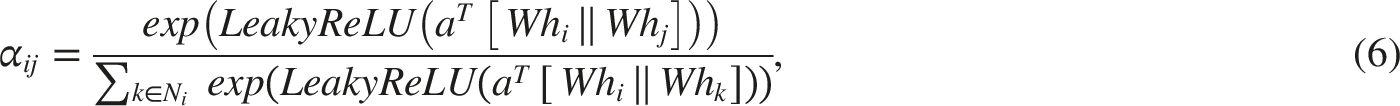
The graph attention mechanism employs 
The node features are updated by weighted aggregation of neighboring node representations, where the weights are given by the learned attention coefficients. The final output feature of node i in a single-head Graph Attention Layer can be described as follows:
Directional-aware graph attention network
Graph attention is formulated as a function
Traditional graph attention networks (GAT) Velickovic et al.
31
; Beaini et al.
40
typically introduce a directional vector
To address the above limitations, we propose a novel Directional-Aware Graph Attention Network (DAGAN). Instead of relying solely on the positional difference vector, DAGAN defines a directional edge embedding vector
Directional edge embedding
However, this vector is simply defined as the positional difference between nodes, i.e., 
Direction-aware attention
The edge embedding 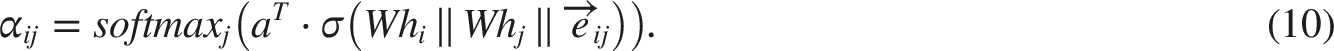
The representation of node 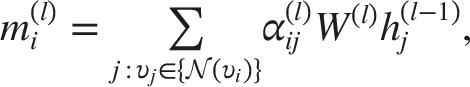

The node update rule for layer 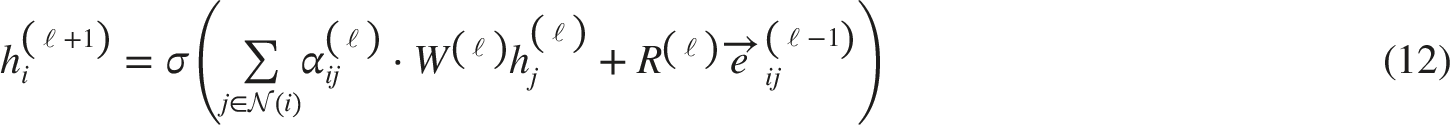
Principle of DAGAN
We proposed DAGAN as shown in Figure 2, which employs two distinct aggregation matrices that assign different weights to nodes within a neighborhood, the directional average matrix The DAGAN framework.

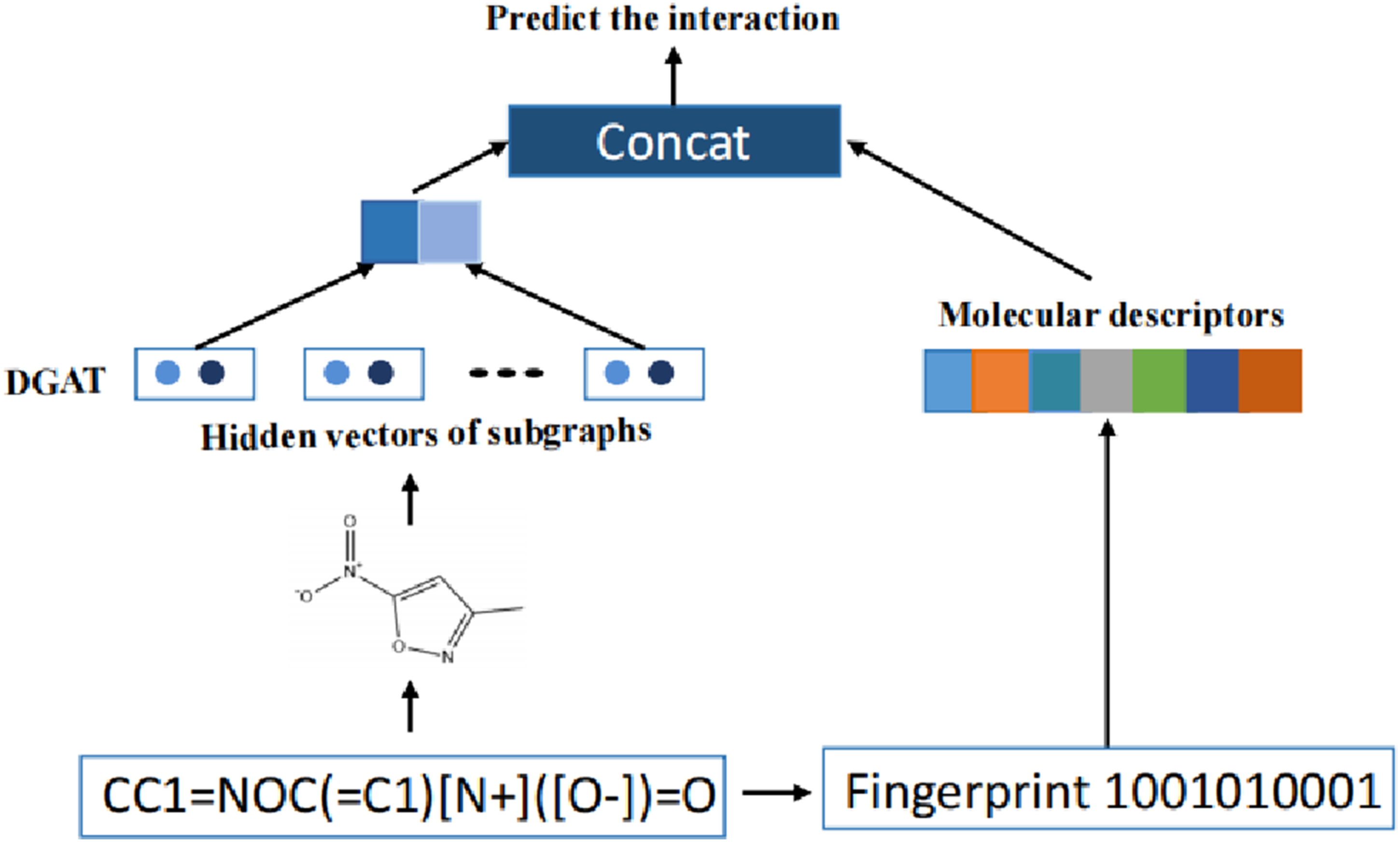
For message passing, DAGAN regards the 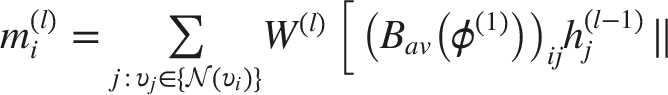
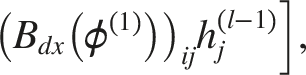
Model training
We take the Directional-Aware Graph Attention Network (DAGAN), a model proposed in this study, as the core model for comparison, aiming to validate its superior performance over traditional machine learning methods. For the task of predicting explosive properties, we adopt the Adam optimizer, which adaptively adjusts the learning rate based on gradient variations during the training process.
For the hyperparameters in the DAGAN model, a grid search method is used to explore the hyperparameter space. The search range for the L2 regularization coefficient is set to [0.0001, 0.01] with a step size of 0.0005. The regularization coefficient range is set to [0.005, 0.1] with a step size of 0.005, and the initial learning rate maximum range is set to [0.05, 0.2] with a step size of 0.05. This hyperparameter setting helps improve the stability and convergence speed of model training, while also preventing the model from getting stuck in local optima.
The total number of training epochs is set to 100, with a batch size of 64 and a learning rate of 0.0001. The model is implemented using the PyTorch deep learning framework and trained on a single NVIDIA GeForce RTX 3090 GPU. The model is trained on the training set and then evaluated on the validation set under different hyperparameter combinations to assess the model’s performance. The model with the best result is tested on the test set, and the average of 5 test runs is used as the final result.
Results and discussion
Comparison for predicting detonation heat
We compare the performance of different machine learning models combined with fingerprint features for predicting the detonation heat of energetic materials. The models compared include SVM, RF, XGBoost, and DAGAN, while considering four types of fingerprint features: MACCS, Daylight, ECFP, and TopoFP. The evaluation metrics used are MAE, RMSE, and Pearson Correlation Coefficient R2 to assess the prediction accuracy of the models.
Comparison for predicting detonation heat(cal/g).
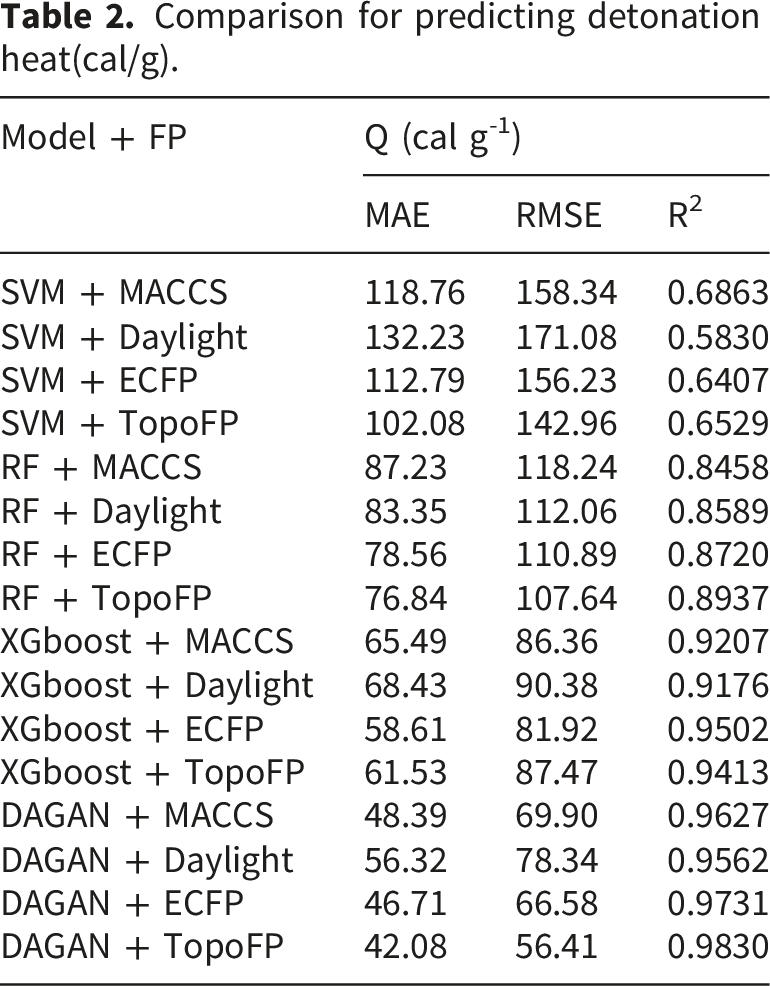
The analysis indicates that DAGAN model performs the best when combined with the TopoFP feature, achieving the lowest MAE and RMSE values, as well as the highest R2 value, demonstrating its superior accuracy in predicting detonation heat. In contrast, the SVM model shows poorer prediction performance when combined with the Daylight fingerprint feature. The XGBoost model performs steadily across all evaluation metrics, particularly when combined with the ECFP and TopoFP fingerprint features. Overall, the results suggest that the DAGAN model offers high prediction accuracy for the task of predicting the detonation heat of energetic materials. Overall, the DAGAN algorithm consistently outperforms the SVM and RF algorithms across all four fingerprint features in detonation heat prediction, while the XGBoost algorithm performs between the two. The DAGAN + TopoFP combination achieves the best results across all three evaluation metrics, indicating its higher accuracy in the detonation heat prediction task.
Comparison for predicting detonation velocity
Comparison for predicting detonation velocity(km/s).
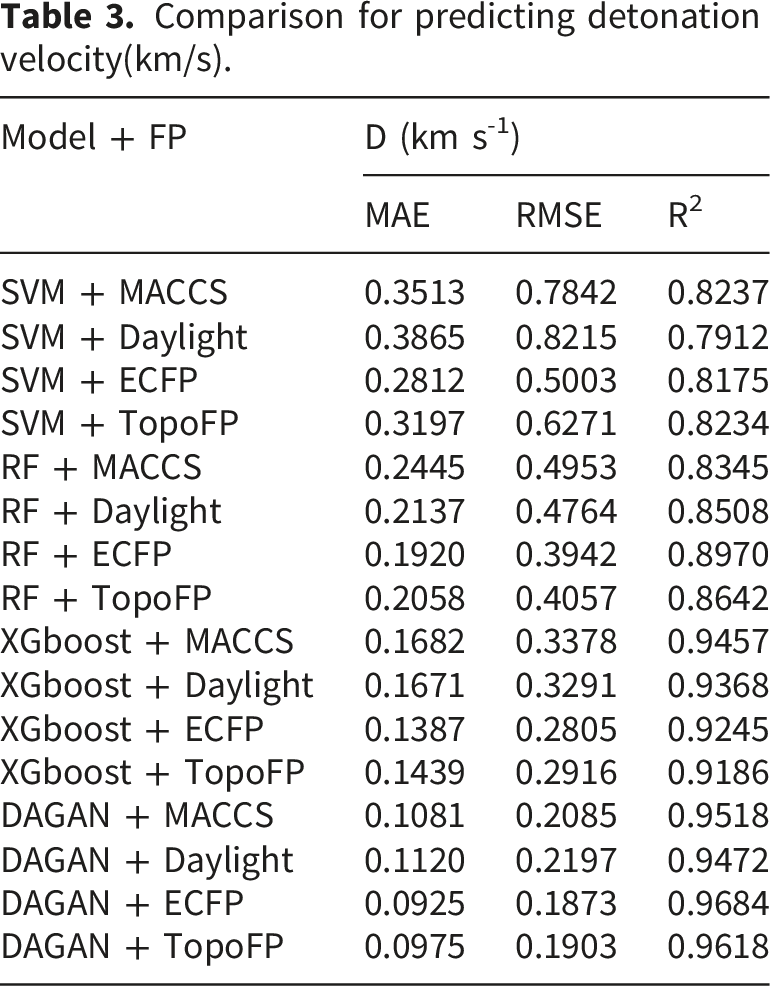
The analysis reveals that the DAGAN model performs most effectively when combined with the ECFP fingerprint feature, achieving the lowest MAE and RMSE values, as well as the highest
Comparison for predicting detonation pressure
Comparison for predicting detonation pressure(GPa).
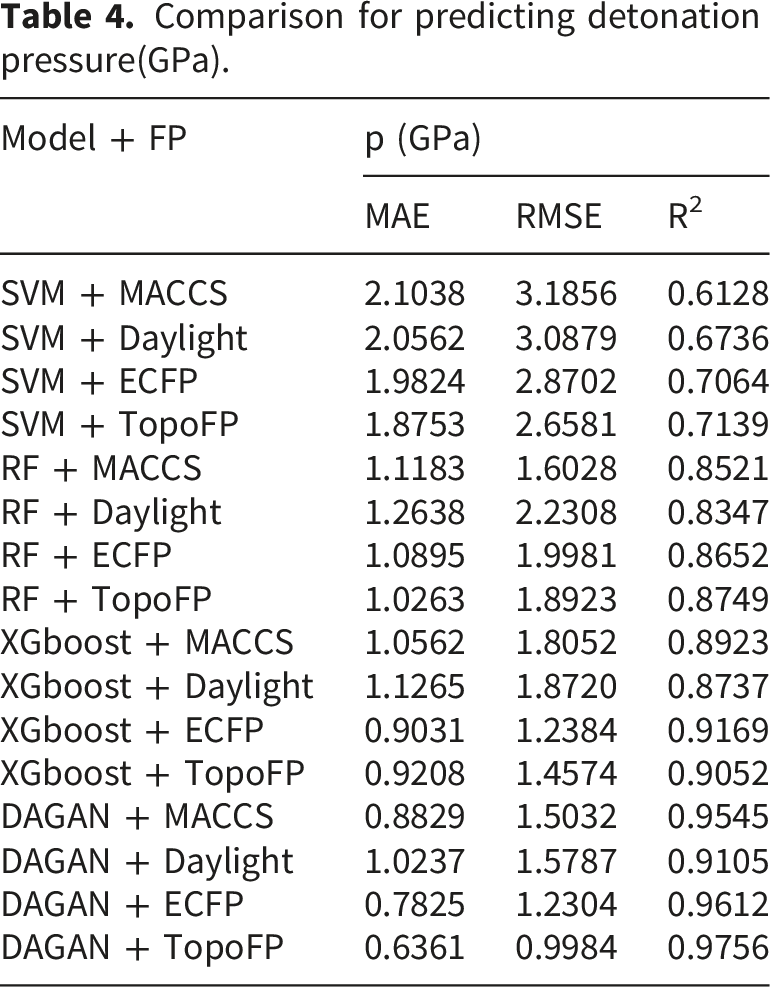
The performance ranking of the models on the prediction set is as follows: SVM < RF < XGBoost < DAGAN, indicating that DAGAN is the most effective model for training and prediction. The attention mechanism enables the model to capture the relationships between atoms and bonds within a molecule, providing both local and global representations. This capability is critical for accurately predicting the properties of energetic materials.
SVM classifies the sample space by finding an optimal hyperplane; however, molecular fingerprint vectors may suffer from information loss—particularly for complex molecular structures—since their fixed-length representation cannot fully capture all molecular details. RF composed of multiple decision trees, builds each tree using random sampling and feature selection from the training data. This ensemble approach helps reduce the risk of overfitting and provides robustness to noise in the data. Additionally, RF excels at feature importance analysis, which aids in identifying the molecular features most critical for property prediction. Nonetheless, descriptors based solely on molecular fingerprints still fall short in capturing the local structural information within molecules, limiting their effectiveness in modeling intricate structure–property relationships.
XGBoost ranks just behind the top-performing models. While it demonstrates strong capabilities in feature extraction from molecular data, it lacks the ability to capture correlations between data points, which affects its overall prediction performance. In the context of predicting the properties of energetic materials, XGBoost can identify some local features effectively. However, it falls short in integrating global structural information and modeling complex molecular interactions, limiting its accuracy in capturing the full behavior of molecular systems.
Model performance analyze
To intuitively analyze the gap between the predicted values and the experimental values, scatter plots were drawn based on the DAGAN model’s predictions, comparing the predicted and actual values of detonation heat, detonation velocity, and detonation pressure using four different molecular fingerprint combinations, as shown in Figure 3. It can be observed that the data points from both the training and testing sets are closely clustered around the line y = x, indicating that the DAGAN model’s predictions are increasingly close to the true experimental values. The learning curve can be used to evaluate the model’s behavior as the number of training changes, providing insights into its learning dynamics and generalization capability. The performance of DAGAN with different fingerprints. The left panel shows the parity plots of the DAGAN model on the test dataset, illustrating the relationship between the predicted values and the equation-calculated values for explosive heat, detonation velocity, and detonation pressure. The right panel presents the training loss curves of the model for explosive heat, detonation velocity, and detonation pressure during the training process.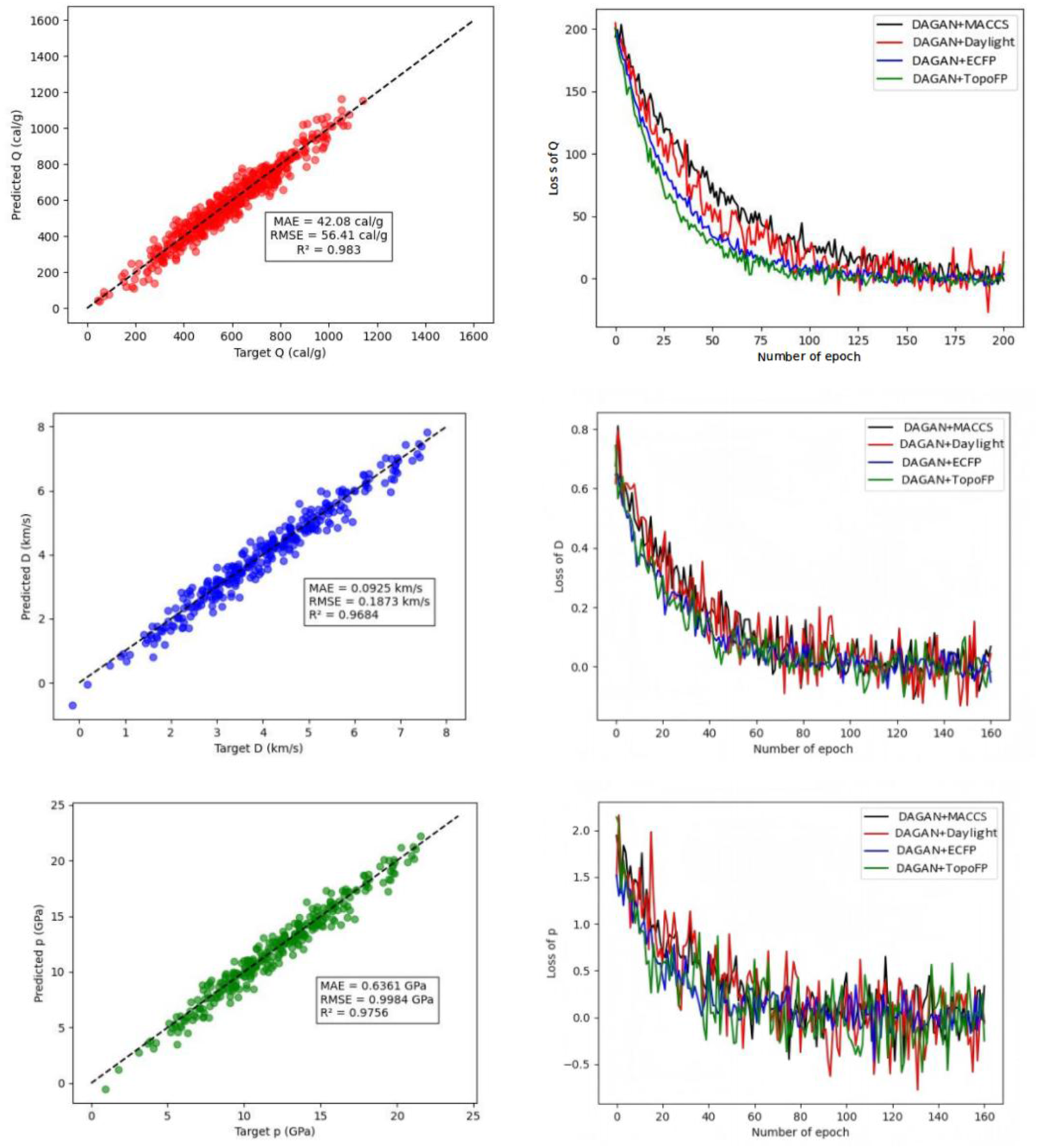
This difference may be attributed to the value distributions of the three properties, as the model exhibits a relatively balanced performance across different ranges, while certain intervals contain fewer data points. The DAGAN model demonstrates strong performance with no signs of overfitting or underfitting. As the number of training samples increases, the improvement in accuracy becomes marginal, indicating that the current dataset size is sufficient to meet the model’s learning requirements. DAGAN is capable of fully preserving the structural information of molecules, including atom types, bond types, and the spatial arrangement of atoms. Unlike traditional machine learning methods based on molecular fingerprints, it does not suffer from the loss of critical structural details due to predefined descriptors or fixed-length vector representations. In energetic materials, even subtle structural variations—such as the position of functional groups or slight changes in bond lengths—can have a significant impact on their properties. DAGAN can accurately capture these fine-grained differences and effectively reflect them in its prediction results, making it particularly well-suited for tasks requiring high structural sensitivity.
The scatter plots in Figure 3 illustrate the optimized prediction results of the DAGAN model on the test sets for the three properties. Among them, the highest predictive performance is achieved for Q (R2 = 0.9830), followed by p (R2 = 0.9756) and D (R2 = 0.9618). This performance discrepancy may be attributed to differences in the numerical distributions of the three properties, as Q exhibits a relatively balanced distribution across different value intervals, whereas D and p have fewer data points in certain ranges. In addition, Figure 3 also presents the training and validation loss curves for the three properties during the training process. After introducing the DAGAN model, the loss values of both the training and validation sets decrease at a faster rate, and the required number of training iterations is reduced, thereby shortening the overall training time.
Based on the experimental results, the DAGAN model consistently outperforms other methods in regression tasks such as the prediction of density, formation energy, detonation heat, detonation pressure, and detonation velocity. Lower error metrics—such as MAE and RMSE alongside higher accuracy and Pearson correlation coefficients, indicate that DAGAN can more precisely predict the molecular properties of energetic materials. This superior performance is attributed to DAGAN’s comprehensive understanding of molecular structure and its effective information propagation mechanism. These strengths enable the model to learn complex relationships between molecular structures and their properties more accurately and efficiently than traditional methods.
In contrast, traditional and conventional machine learning methods generally exhibit weaker interpretability, making it difficult to intuitively understand the internal relationships between model predictions and molecular structures. DAGAN, however, enables parallel computation of atom–neighbor interactions, can handle nodes with varying degrees, and assigns different attention weights to neigh-boring atoms. This not only validates the rationality of the model’s predictions but also provides an intuitive foundation for understanding the structure–property relationships in energetic materials.
Conclusions
This paper presents the DAGAN prediction model for energetic materials, focusing on their molecular structures and associated explosive properties. The model’s network architecture, feature representation of nodes and edges, and training algorithm are designed. The model is then trained and optimized through experiments, calculating features such as the distribution of atom types around nodes and the topological environment of chemical bonds to enrich the initial feature representations of nodes and edges. This approach effectively captures the inherent characteristics of molecular structures. Finally, the DAGAN model is compared with SVM, RF, and XGBoost, and the model’s prediction results are interpreted.
Compared to traditional machine learning methods, DAGAN excels in capturing both local and global interactions between atoms within a molecule, avoiding the information loss issues inherent in conventional feature extraction methods. The DAGAN method demonstrates significant superiority over other machine learning approaches in predicting the properties of energetic materials, offering a more powerful and accurate prediction tool for research and development in this field. Looking forward, further exploration into how to fully leverage the advantages of DAGAN, as well as improvements in model architecture and training algorithms, will be essential to meet the growing challenges and demands in the energetic materials domain. In the future, We will test on the same dataset with a large language model to achieve better experimental results.
Footnotes
Acknowledgements
This research was funded by the Science and Technology Tackling in Henan Province (No 252102240011, 252102210166) and the Special Research Program for Basic and Frontier Technologies of Nanyang City (No 23JCQY2023).
Author contributions
Juncheng Yang conceptualized the workflow of this paper. Xiaoyang Zhao conducted the formal analysis required. Shuxia Li wrote the original draft. Shiquan Li contributed to the data interpretation and discussion of the results. All authors reviewed the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Science and Technology Tackling in Henan Province (252102240011); (252102210166), and the Special Research Program for Basic and Frontier Technologies of Nanyang City (23JCQY2023).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.