
Abstract
Cancer survival prediction is crucial for clinical decision-making and personalized treatment planning. The joint analysis of pathological images and genomic profiles provides complementary information at both histological and molecular levels, offering a more comprehensive foundation for patient prognosis assessment. However, existing multimodal survival prediction methods face two major challenges: (1) How to efficiently capture global dependencies in high-dimensional, long sequence features while maintaining linear complexity? (2) How to fully preserve and utilize the inherent valuable information of each modality while achieving cross-modal interaction? To address these challenges, we propose MCAMamba, a multimodal method with bidirectional cross-attention and a state space model for cancer survival prediction. This method employs a parallel encoder–decoder architecture, leveraging Mamba’s efficient long sequence modeling capabilities to capture global dependencies and discriminative features within each modality. Meanwhile, a Bidirectional Cross-Attention module is integrated into the framework to achieve semantic alignment and complementary information exchange across modalities, enhancing the prediction of patient survival risk. Experimental results on four public TCGA cancer datasets (BLCA, BRCA, UCEC, and LUAD) demonstrate that MCAMamba significantly outperforms existing methods in predictive performance. The c-index improves by 2.47%-17.9%, validating the superior performance of the method in multimodal cancer survival prediction.
1. Introduction
Cancer, characterized by its high incidence and mortality rates, has become one of the most serious public health challenges worldwide. 1 Accurate prediction of patient survival is crucial for clinical decision-making, as it enables early risk stratification and supports the development of personalized treatment plans. However, due to the complex nature of cancer, relying solely on single-modality data often fails to comprehensively reflect a patient’s true condition. As cornerstones of modern oncology, pathology and genomics provide unique insights into cancer research at the macroscopic morphological and microscopic molecular levels, respectively. Pathological images, particularly whole-slide images (WSIs) at gigapixel resolution, capture spatial information such as tumor architecture, spatial heterogeneity, and the tumor microenvironment, whereas genomic profiles reveal underlying molecular mechanisms. These two modalities are inherently complementary.2–4 Therefore, obtaining feature representations that are both discriminative and biologically interpretable, and achieving efficient integration of information between pathological images and genomic profiles, has become a major research focus and challenge in cancer survival prediction.
Due to the extremely high resolution of WSIs, survival prediction tasks commonly employ Multiple Instance Learning (MIL) methods. In MIL, a WSI is first divided into multiple patches and encoded into low-dimensional feature representations using pre-trained models.5–7 These instance features are then aggregated into a bag for downstream tasks. This process formulates WSI feature extraction as a long-sequence modeling problem to capture inter-instance correlations and global contextual information for discriminative feature learning. Although Transformer-based methods8–10 effectively capture global dependencies in long sequences, their computational complexity has quadratic growth with sequence length, creating a major bottleneck in high-dimensional sequence analysis. To address this limitation, the Selective Space State Sequence Model (Mamba) 11 was introduced as an efficient alternative for long-sequence modeling. Existing research demonstrates that Mamba achieves comparable or superior performance to Transformers across multiple tasks while requiring only half the parameters.12,13 For example, MamMIL 14 integrates Mamba into the MIL framework for WSI analysis, thereby enabling effective modeling of global instance dependencies with linear computational complexity. These findings provide the theoretical foundation and primary motivation for introducing Mamba into multimodal survival prediction tasks in this work.
In recent years, with advances in multimodal learning methods, an increasing number of studies15–19 have integrated pathological images with genomic data for cancer survival analysis, significantly improving the accuracy of patient survival prediction. Existing multimodal fusion methods can be broadly categorized into two types. The first category includes direct feature fusion methods, which integrate modalities at the feature level through techniques such as concatenation20,21 and bilinear pooling.16,22,23 Although these methods generate joint feature representations, they often overlook potential interactions between modalities, resulting in limited representational capacity of the fused features. The second category comprises cross-modal interaction methods, which introduce attention mechanisms to guide and align information across modalities, thereby capturing latent dependencies between pathological and genomic features. Notable works include, Chen et al. 24 proposed the Multimodal Co-Attention Transformer (MCAT) framework, which facilitates interaction between pathological and genomic features through a genome-guided co-attention mechanism. Jaume et al. 17 proposed the SurvPath model, which employs sparse attention to simulate interactions between genomic pathways and histological patch tokens, thereby enhancing feature complementarity. Although these methods have made notable progress in improving predictive performance, they primarily emphasize shared intermodal features while failing to fully preserve and utilize the inherent valuable information, leading to the loss of critical intra-modal insights. Therefore, future research should focus on effectively modeling cross-modal interactions while preserving informative intra-modal representations, thereby fully leveraging the complementary strengths of multimodal data to improve cancer survival prediction.
Based on the above observations, we propose MCAMamba, a multimodal method with Bidirectional Cross-Attention and a State Space Model for cancer survival prediction. The proposed method is designed to efficiently model intra-modal representations of pathological images and genomic profiles while exploring cross-modal correlations and complementary information. Specifically, MCAMamba adopts a parallel encoder–decoder architecture. Leveraging Mamba’s efficient long sequence modeling capability, the framework captures intra-modal feature representations for the pathological and genomic modalities. Subsequently, A Bidirectional Cross-Attention (BCA) module is further embedded into the architecture to explicitly model cross-modal correlations and facilitate bidirectional interaction and semantic alignment between the two modalities. Finally, Self-Attention Pooling (SAP) aggregates global representations of each modality for patient survival risk prediction. The main contributions of this work are summarized as follows: (1) We propose MCAMamba, a multimodal method based on bidirectional cross-attention mechanisms and state space model for cancer survival prediction. This method enables deep interaction and fusion of pathological images and genomic profiles, significantly improving survival prediction accuracy. (2) Leveraging Mamba’s efficient long sequence modeling capability, we construct a cross-symmetric encoder–decoder architecture to fully capture global dependencies and discriminative features within each modality. (3) We introduce a Bidirectional Cross-Attention module, embedding within the encoder–decoder architecture. This module explicitly models intrinsic correlations between pathological images and genomic data, enabling bidirectional information guidance and efficient transfer of complementary features. (4) Extensive experiments on four public TCGA cancer datasets (BLCA, BRCA, UCEC, and LUAD) evaluate the effectiveness of our proposed model. The results demonstrate that our model significantly outperforms existing methods in predictive performance.
The rest of this paper is organized as follows. Section 2 primarily discusses related works. Section 3 introduces the basic concepts of state space model, Mamba, and survival prediction, followed by a detailed description of the components of the MCAMamba framework. Section 4 introduces the cancer datasets, experimental setup, experimental results and model interpretability. Section 5, provides a discussion of the proposed method. Finally, Section 6 concludes the paper.
2. Related work
2.1. Survival prediction from single modality
Pathological images and genomic profiles are increasingly recognized as crucial prognostic indicators for cancer, demonstrating substantial potential in survival prediction tasks. In pathological images analysis, survival prediction research is primarily conducted within the MIL framework. Lee et al. 25 proposed DeepSets, pioneering the integration of set-based learning concepts into pathological image feature modeling. This method enables models to learn global representations from unordered patch inputs, laying the foundation for subsequent MIL studies. Ilse et al. 26 developed AttentionMIL, which achieves adaptive aggregation of patch-level features through an attention-weighted mechanism, significantly enhancing the model’s discriminative power and interpretability. Shao et al. 8 introduced TransMIL, incorporating the Transformer architecture into the MIL framework. By explicitly capturing global dependencies and correlations among patches via a self-attention mechanism, it strengthens the model’s ability to learn long-range dependencies. Yao et al. 27 proposed DeepAttnMISL, employing an attention-guided MIL pooling strategy to adaptively weight patch features from WSIs at the patient level for cancer survival analysis, thereby further improving model interpretability. In genomics, features are typically represented as one-dimensional measurements (1×1 vectors). Feature modeling can be achieved using methods such as Multi-Layer Perceptron (MLP), 28 Self-Normalizing Networks (SNN), 29 and DeepSurv. 30 Among these, DeepSurv serves as a foundational model for deep neural network–based survival prediction. By integrating the Cox proportional hazards model with deep neural networks, it enables the learning of nonlinear risk functions, enhancing the expressive power and flexibility of survival prediction. Although these single modality methods have achieved substantial progress in feature extraction and risk modeling, they characterize tumor features only from a single dimension. This limitation hinders a comprehensive understanding of the complex mechanisms underlying cancer development and progression, constraining further improvements in predictive performance.
2.2. Survival prediction from multiple modalities
To overcome the limitations of single modality methods, increasing research in recent years has explored the joint modeling of pathological images and genomic profiles. This line of work aims to fully leverage the complementary information within multimodal datasets, thereby enhancing the accuracy of cancer survival prediction and improving model generalization capability. Cao et al. 16 proposed PORPOISE, introducing bilinear pooling into multimodal fusion. By modeling higher-order interactions, it explicitly captures genotype–phenotype associations; however, its fusion strategy remains globally biased, with insufficient emphasis on locally critical regions. To further refine cross-modal alignment precision, Xu et al. 31 introduced the MOTCat framework, which establishes fine-grained correspondences between pathological and genomic features through optimal transport matching and global structural consistency constraints, yielding more accurate and reliable cross-modal mappings. Liu et al. 32 proposed the Mutual-Guided Cross-Modal Transformer (MGCT), which simulates genotype–phenotype interactions within the tumor microenvironment to enhance the consistency and contextual relevance of cross-modal representations. Zhou and Chen 19 proposed the Cross-modal Translation and Alignment (CMTA) framework to explore intrinsic cross-modal correlations and extract latent complementary information. Yang et al. 33 introduced MMsurv, which combines bilinear pooling with a transformer architecture to effectively integrate diverse data types, thereby strengthening the feature expression capability of predictive models. Although the aforementioned methods have achieved notable results in multimodal survival analysis, the cross-modal interaction process may still lead to the loss or redundancy of inherent valuable information.34,35 To address this issue, we propose an improved cross-modal fusion strategy based on the SAMambar, 36 aiming to more comprehensively explore potential cross-modal correlations and complementarities, thereby enhancing the accuracy of survival analysis.
2.3. Mamba
In recent years, State Space Models (SSMs) have demonstrated significant advantages in handling dynamic systems and modeling long-range dependencies, gradually gaining widespread application in medical image analysis and multimodal learning. Yang et al. 37 proposed the MambaMIL model, which integrates Mamba into a MIL framework. By employing a sequence reordering strategy, it captures long-range dependencies among dispersed instances. This method effectively mitigates overfitting and computational overhead while maintaining linear complexity, thereby enhancing the model’s ability to capture key discriminative features. Dang et al. 38 proposed the LoG-VMamba model, which further integrates local and global feature modeling. It outperformed CNN and Transformer-based baselines in various 2D and 3D medical image segmentation tasks, validating Mamba’s potential for modeling long-range dependencies and spatial consistency. For survival prediction, Chen et al. 39 proposed SurvMamba, a multi-granularity and multimodal interaction model. This model comprises two modules: Hierarchical Interaction Mamba (HIM) and Interaction Fusion Mamba (IFM). HIM captures correlations among features at different levels of granularity, while IFM facilitates the fusion of cross-modal interactions. This dual-module architecture enhances feature representation at both fine-grained and global levels, thereby significantly improving the accuracy and efficiency of multimodal survival prediction. Song et al. 40 proposed The DSCASurv framework, which combines the local feature extraction capability of convolutional layers with the long-range dependency modeling ability of Mamba. This method captures intrinsic correlations between pathology and genomics during cross-modal fusion and alignment, further improving multimodal survival prediction performance. In summary, Mamba provides a novel paradigm for modeling complex medical data through its exceptional long sequence modeling capability and structural flexibility. We construct a parallel encoder-decoder architecture based on Mamba to fully capture global dependencies and discriminative features within modalities, thereby enhancing the expressive power of multimodal representations and improving survival prediction performance.
3. Method
In this section, we systematically introduce the proposed multimodal survival prediction framework, MCAMamba, as shown in Figure 1. Section 3.1 briefly reviews the fundamentals of state space models and their variant, Mamba. Section 3.2 introduces the survival prediction objective. Section 3.3 presents the data processing and feature extraction methods for both modalities. Section 3.4 describes the pathology encoder and genomics encoder. Section 3.5 details the Bidirectional Cross-Attention module. 3.6 describes the pathology decoder and genomics decoder. Section 3.7 discusses feature fusion and survival prediction. Framework of the MCAMamba method. (1) Segment the WSI into patches and use the pre-trained feature extractor CTransPath to obtain representative pathological features. (2) Perform gene enrichment analysis on genomic profiles to identify biologically enriched pathways and generate pathway level features. (3) Construct a parallel encoder-decoder architecture, embedding a Bidirectional Cross-Attention module to explore intrinsic cross-modal correlations and transmit latent cross-modal information. Finally, aggregate features within each modality using SAP and feed the fused features into a MLP for final survival prediction.
3.1. Preliminaries


To adapt this model for deep learning systems, the Structured State Space Sequence Model (S4) discretizes the continuous system. Using the Zero Order Hold method and introducing a time step Δ, the continuous parameters A and B are transformed into discrete parameters


After discretization, the discrete version with step size Δ can be expressed as:







This mechanism allows the model to dynamically adjust the state evolution process according to different inputs, thereby more effectively modeling long-range dependency information with linear time complexity. In this study, Mamba is employed to model genomic pathway sequences and pathological patch sequences, providing efficient representations with long-range dependency structures for subsequent multimodal fusion.
3.2. Survival prediction objective
In multimodal survival analysis tasks, each patient’s sample data is represented as a quadruple

Since accurately predicting a patient’s exact survival time is challenging, we employ discrete-time modeling to estimate the probability of surviving beyond a set of discrete time points. Based on the hazard function, the cumulative survival function

3.3. Data processing and feature extraction
Each WSI typically contains thousands or even tens of thousands of patches, with dense distributions and highly redundant features. Directly processing all patches would incur extremely high computational costs and increase model instability. To address this issue, we introduce a Patch Clustering Layer (PCL)
36
to extract representative key regions from the extensive patch feature set. Specifically, the PCL module maintains K trainable cluster centers

Through the aforementioned process, K representative cluster features are selected from the original large-scale patch collection to construct patch feature sequences for pathological images. The PCL module compresses the original high-dimensional patch sequence into a representative patch feature sequence of length K, effectively reducing computational complexity for the subsequent pathology encoder and Bidirectional Cross-Attention Module. Ultimately, each patient’s pathological features can be formally represented as
Subsequently, we employed SNN
29
to perform embedding mapping on the filtered pathway features, uniformly projecting pathway features of different dimensions into a low-dimensional space to obtain more discriminative pathway representations. Ultimately, each patient’s genomic features are represented as:
3.4. Pathology encoder and genomics encoder
Given that both pathological images and genomic profiles exhibit characteristics such as high dimensionality and long-range dependencies,15,32 we leverage Mamba’s efficient long sequence modeling capability to construct parallel encoder–decoder architectures. This design captures global dependencies and discriminative features within each modality. Specifically, the structure of the pathology decoder aligns with that of the genomics encoder, while the genomics decoder mirrors the structure of the pathology encoder.
Structural diagram of SR-Mamba.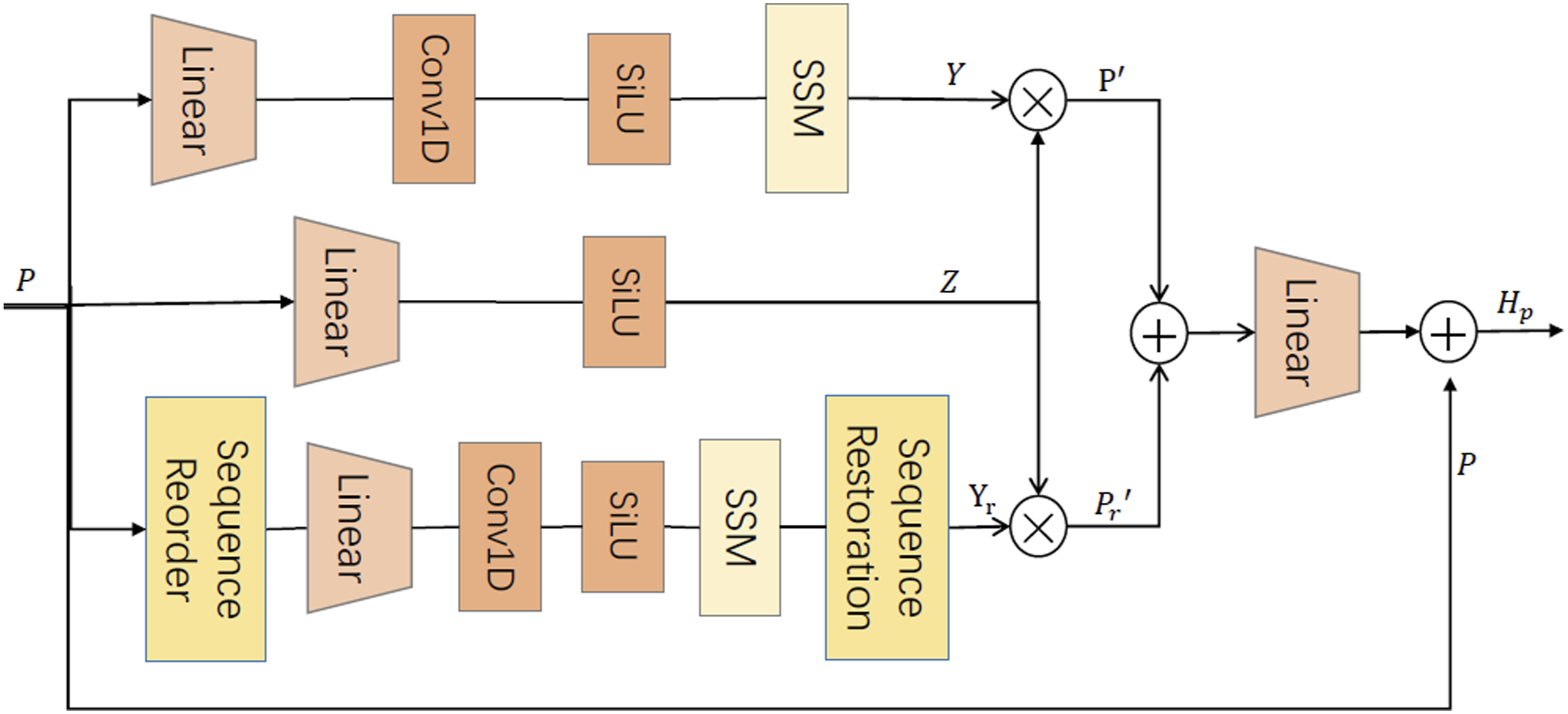
To further elucidate the working mechanism of SR-Mamba, its specific implementation steps are detailed below. (1) Temporal Modeling Branch. This branch preserves the original sequence order P. It captures long-range dependencies using a casual convolutional layer and a state space model, producing the output representation Y:

To achieve dynamic modulation, we generate a gating signal from the input sequence:

Subsequently, the generated gating signal Z is used to perform weighted modulation on Y, yielding the discriminative features for the temporal branch: (2) Spatial Rearrangement Branch. This branch reconstructs the sequence


Consistent with the Temporal Modeling Branch, we similarly use the generated gating signal Z to weight the reordered output features


The dual-branch architecture of SR-Mamba preserves cross-regional contextual dependencies while maintaining the spatial consistency of local organizational structures. The final pathological feature
Structural diagram of Mamba.
The specific process is as follows: Given a pathway feature sequence

Subsequently, the state space parameters are dynamically generated based on x' and fed into the SSM to model cross-channel long-range dependencies:

The output feature Y is then weighted by the gate signal z:

Finally, the genomic modality output representation is obtained through a linear transformation:

This genomics encoder efficiently captures cross-pathway long-range dependencies while preserving the discriminative features of local pathways features. We denote the genomic feature output
3.5. Bidirectional cross-attention module
Pathological images and genomic profiles reflect tumor characteristics at the levels of histological morphology and molecular mechanisms, respectively. During cancer progression, morphological alterations in specific regions of pathological images are often accompanied by abnormal expression of related genes.43,44 Based on this observation, we propose a Bidirectional Cross-Attention module to explicitly model bidirectional dependencies between pathological and genomic features. This module establishes mutually guided attention pathways to achieve dynamic semantic alignment and feature complementarity between the two modalities. It further uncovers intrinsic correlations between pathology and genomics, enabling mutual guidance and bidirectional transmission of cross-modal information.
Specifically, we first concatenate pathological features
Within the BCA, each modality serves as the query vector, while the other modality’s features act as key and value vectors, enabling bidirectional information exchange:


The cross-attention process from pathological modality to genomic modality (Path→Gen) is:

The cross-attention process from the genomic modality to the pathological modality (Gen→Path) is:

Through this bidirectional cross-attention mechanism, the BCA enables two-way information exchange and complementary data transmission between modalities. This allows the pathological modality to focus spatially on regions associated with key pathways, while the genomic modality acquires more contextually meaningful representations, thereby facilitating deep interactions between pathological and genomic characteristics.
3.6. Pathology decoder and genomics decoder
To further map the cross-modal features obtained from bidirectional cross-attention into representations consistent with the target modality’s semantic space, we introduce two modality decoders after the bidirectional cross-modal interaction module. This enables bidirectional information flow and semantic complementarity during the decoding phase, thereby enhancing the alignment and fusion of cross-modal features. Specifically, the pathology decoder takes genome-guided pathological features

Here,
Correspondingly, the genome decoder takes the pathology-feature-guided genomic feature

In the structural design, the pathology decoder adopts the Mamba architecture consistent with the genome encoder, while the gene decoder employs the SR-Mamba architecture aligned with the pathology encoder. This ensures symmetric representational capacity in both Gen→Path and Path→Gen directions, reducing directional bias and enhancing alignment stability. The decoder outputs
3.7. Feature fusion and survival prediction
To further focus on key tissue regions and genomic pathways strongly associated with disease, we feed the decoded pathological features


To optimize model training, we adopt the Negative Log-Likelihood loss with censoring, a widely used objective function in survival analysis. This loss maximizes the likelihood of patient survival while appropriately handling censored data, thereby improving predictive accuracy. The loss function is defined as follows:

4. Experimental results and analysis
In this section, we conduct extensive experiments on four public TCGA datasets to evaluate the effectiveness of our method. First, we introduce the datasets, evaluation metrics and implementation details used in our study. Next, we compared our method with several state-of-the-art methods to demonstrate its superiority. Then, we perform ablation studies to evaluate the contribution of each key module in our method. Finally, we provide visual statistical analysis and interpretation to verify the validity and reliability of the proposed method in survival prediction.
4.1. Datasets and settings
Dataset statistics across different cancer types.
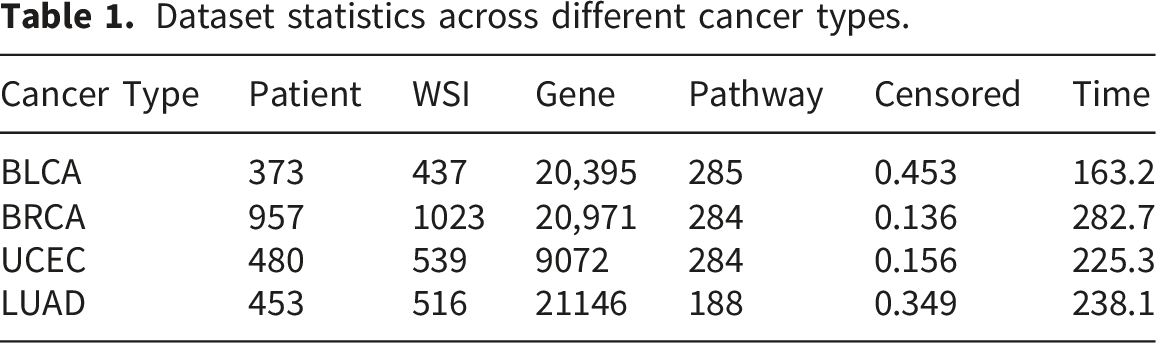
4.2. Comparison with state-of-the-art methods
To comprehensively evaluate the proposed multimodal survival prediction method MCAMamba, we compared it with a variety of existing methods using a 5-fold cross-validation strategy on four TCGA cancer datasets (BLCA, BRCA, UCEC, and LUAD). The comparison included both single-modal methods and multi-modal methods. (1) Single-modal methods.
To assess the predictive capability of each individual modality, we conducted single-modal comparison experiments on pathological images and genomic data separately. For the genomic modality, we selected three representative baseline models: MLP,
28
SNN,
29
and DeepSurv.
30
For pathological images, we compared several representative MIL methods, including DeepSets,
25
AttentionMIL,
26
DeepAttnMISL,
27
and TransMIL.
8
As shown in Table 2, MCAMamba outperformed all single-modality methods on the four datasets, achieving optimal survival prediction performance. Compared with genomic unimodal methods, the average c-index improvement ranged from 15.4% to 18.9%; compared with pathology unimodal methods, improvements ranged from 18.2% to 38.1%. These results demonstrate that relying solely on a single modality fails to capture the complex phenotype–molecular dependencies in cancer. Whereas integrating histological and genomic information substantially, MCAMamba markedly enhances survival prediction performance and model generalization. (2) Multi-modal methods. Performance comparison of different methods across four TCGA datasets, with the best performer highlighted in bold.

To validate the effectiveness of our model, we compared MCAMamba with seven state-of-the-art multimodal survival prediction models that share the same task definition as this paper and provide publicly available implementations, including: MCAT, 24 PORPOISE, 16 MGCT, 32 MOTCat, 31 CMTA, 19 SAMamba, 36 and SurMoE. 45 As shown in Table 2, MCAMamba achieved the best performance across all comparison methods. Specifically, it outperformed all other models on the BLCA (0.682), BRCA (0.676), UCEC (0.762), and LUAD (0.692) datasets. Compared with existing multimodal methods, MCAMamba achieved average c-index improvements ranging from 2.47% to 17.9%, demonstrating the model’s superiority in multimodal survival prediction. Notably, compared with SAMamba the structurally closest multimodal survival prediction method MCAMamba achieved higher c-index values across four datasets. These results indicate that the proposed MCAMamba effectively embeds a Bidirectional Cross-Attention module within a parallel encoder–decoder architecture to enhance cross-modal alignment and preserve intra-modal features, thereby improving the model’s ability to predict patient survival.
4.3. Ablation studies
To evaluate the contribution of each key module in our method to survival prediction performance, we conducted ablation experiments on four TCGA cancer datasets. We first verified the validity of each module within the model. Then, we explored the impact of the Mamba and the number of cluster centers in PCL modules on model performance.
(1) Impacts of Patch Clustering Layer: To validate the effectiveness of PCL clustering phenotype, we conducted experimental evaluations by removing this module from the MCAMamba (w/o PCL). Removing PCL resulted in performance declines across all datasets, with the most significant decreases in BLCA (−3.96%) and UCEC (−3.67%). This indicates that the PCL module contributes positively to overall model performance by reducing redundant patches and preserving key regional features. (2) Impacts of SR-Mamba module: To validate the effectiveness of state-space modeling in SR-Mamba, we replaced the SR-Mamba component in the pathology image encoder with the standard Mamba architecture (Mamba). Replacing SR-Mamba with standard Mamba led to substantial performance degradation across all datasets: BLCA decreased by 5.73%, BRCA by 5.29%, UCEC by 4.09%, and LUAD by 3.12%. This demonstrates that the SR-Mamba module more effectively captures spatial structures and cross-regional dependencies in pathological images, thereby enhancing the representational capability of the pathological modality. (3) Impacts of Bidirectional Cross-Attention Module: To validate the interactive effects of BCA, we conducted experimental evaluations by removing this module from the MCAMamba (w/o BCA). Removing BCA caused substantial degradation in cross-modal interaction capability. Specifically, the c-index decreased by 11.2% for BLCA, 11.3% for BRCA, 7.9% for UCEC, and 2.5% for LUAD. These results indicate the pivotal role of the BCA in facilitating mutual guidance and fusion between pathological and genomic features, as well as in capturing cross-modal complementary information. Without this mechanism, the model struggles to effectively integrate correlated features across modalities, resulting in significantly reduced predictive performance. (4) Impacts of Self-Attention Pooling: To validate the interactive effects of SAP, we conducted experimental evaluations by removing this module from the MCAMamba (w/o SAP). Removing SAP led to varying impacts across datasets. UCEC and LUAD showed minor changes (3.67% and 1.61%, respectively), whereas BLCA and BRCA exhibited c-index declines of 7.74% (from 0.682 to 0.633) and 6.28% (from 0.676 to 0.636). This demonstrates that the SAP module enables the model to focus on disease-relevant pathological regions and gene pathways through its adaptive weighting mechanism. When removed, the model’s ability to aggregate local features weakens, leading to diminished risk characterization. Experimental results after removing the following key components, with the complete MCAMamba model highlighted in bold.

Overall, removing or replacing any core module led to performance degradation, with the removal of the BCA producing the most pronounced effect (a maximum decline of 11.3%). These results validate the critical roles of each component in the MCAMamba model: PCL enhances input feature quality, SR-Mamba strengthens spatial dependency modeling, BCA promotes cross-modal feature alignment and complementarity, and SAP improves key feature aggregation. The synergistic interactions among these modules collectively underpin the model’s superior performance in multimodal cancer survival prediction.
The trend of single inference time with respect to different backbone networks. (a) Median latency curve. (b) p95 latency curve. Efficiency and resource usage (K=2048, batch=1).
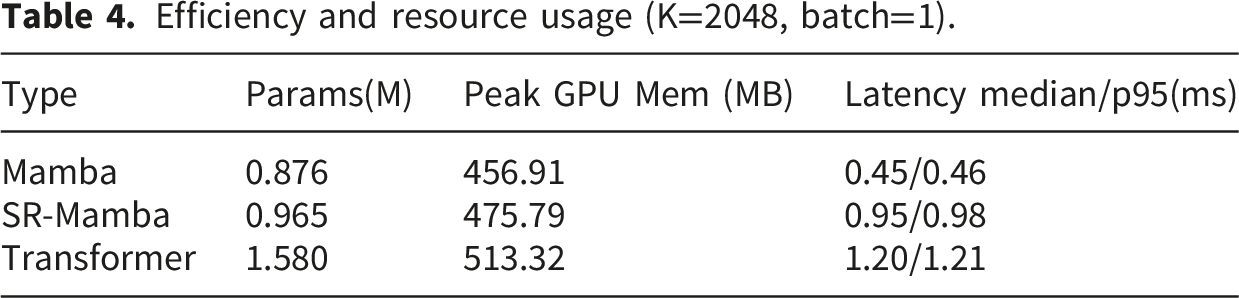
Performance under different numbers of cluster centers K, particularly in terms of the c-index.
4.4. Survival analysis
To evaluate the effectiveness of the MCAMamba model in patient risk stratification, we performed Kaplan-Meier curves based on the model-predicted risk scores, as shown in Figure 6. Specifically, patients were divided into high-risk and low-risk groups based on the median predicted risk score; to ensure a fair comparison, all baseline models employed the same median stratification strategy. The Kaplan-Meier curves were used to estimate the cumulative survival probability of patients in different risk groups. Survival differences between groups were assessed using the log-rank test, with the significance level set to α=0.05 (all p-values in Figure 6 originate from this test). The stratification and separation trends observed in the Kaplan-Meier curves demonstrate that, across the four cancer datasets (BLCA, BRCA, UCEC, and LUAD), MCAMamba exhibits a more pronounced separation between high-risk and low-risk groups than the other two competitive multimodal methods. The log-rank test results indicate statistically significant survival differences between the high-risk and low-risk groups for all datasets. On the BLCA dataset, the log-rank test p-values were 4.3989e-12 (SAMamba), 1.8946e-05 (SurMoE), and 5.4127e-16 (MCAMamba), with MCAMamba demonstrating the most significant intergroup differences. On the BRCA dataset, the p-values were 1.3786e-07 (SAMamba), 1.0723e-06 (SurMoE), and 5.3560e-07 (MCAMamba), all statistically significant. On the UCEC dataset, the p-values were 1.7847e-11 (SAMamba), 1.6692e-05 (SurMoE), and 3.4375e-16 (MCAMamba), again indicating a more pronounced stratification effect for MCAMamba. On the LUAD dataset, the p-values were 3.1704e-09 (SAMamba), 4.3872e-09 (SurMoE), and 5.6824e-17 (MCAMamba), again demonstrating that MCAMamba achieved the most significant survival differences between the high-risk and low-risk groups on this dataset. These results further validate MCAMamba’s ability to effectively distinguish patient cohorts with differing prognostic risks, demonstrating stable and competitive risk stratification capabilities across multimodal survival prediction tasks. Kaplan-Meier curves and log-rank test results.
4.5. Interpretability
To further investigate the interpretability of the model’s predictions, we conducted a visual analysis of the basis of the model’s predictions from three perspectives: pathological regions, genomic pathways, and cross-modal correlations. As shown in Figure 7, at the pathological level, the model mainly attends to regions with abundant stromal components, disrupted glandular structures, and marked cellular atypia in the high-risk case, indicating that its risk assessment relies on morphological cues associated with aggressive growth and disorganized tissue architecture. In contrast, in the low-risk case, the model focuses on regions with more intact glandular structures and more continuous tumor epithelial arrangements, suggesting that it relies more on areas with better differentiation and more stable tissue architecture. These results indicate that the model can capture key histological patterns associated with different prognostic states, thereby providing a pathological basis for risk prediction. At the pathway level, the top pathways in the high-risk case included Autophagy-animal, Phosphatidylinositol signaling system, MAPK signaling pathway, Endocytosis, and Endometrial cancer, reflecting enhanced tumor cell survival, activation of oncogenic signaling, and processes related to invasion and migration. By contrast, the top pathways in the low-risk case mainly included the GnRH signaling pathway, mTOR signaling pathway, Cellular senescence, and TGF-beta signaling pathway. Compared with the high-risk case, these pathways were more dispersed and were generally associated with endocrine regulation, maintenance of cellular state, and growth regulation, rather than with invasion, migration, and stress-survival characteristics. Overall, the model can distinguish distinct pathway patterns across risk groups: high-risk cases are characterized by stronger tumor progression-related pathways, whereas low-risk cases exhibit a more dispersed and relatively regulatory pathway pattern. This observation is consistent with the top-patch analysis, indicating good agreement between the model’s pathway-level and histopathological interpretations. Interpretability and attention visualization.
From the perspective of cross-modal correlations, we selected the same four representative pathways for visualization in both groups: MicroRNAs in cancer 46 (human diseases); the MAPK signaling pathway 47 (signal transduction); focal adhesion 48 (cellular processes); and FoxO signaling pathway 49 (Signal Transduction). These maps depict the distribution of cross-modal attention weights assigned by the model to different pathological regions under the given pathway conditions. All attention values underwent min-max normalization within each WSI (range [0, 1]) to enhance spatial contrast within the same sample. Consequently, color intensity primarily reflects the relative spatial distribution within a sample rather than absolute numerical comparisons between cases. A unified turbo color-map is employed, where warm colors (red/orange) denote relatively high attention weights and cool colors (blue) indicate relatively low weights. Figure 7 reveals distinguishable spatial weight distribution patterns between the two case groups. High-risk cases exhibit more extensive distributions of medium-to-high attention intensity for the MAPK signaling and MicroRNAs in cancer pathways, whereas high-attention regions in low-risk cases are relatively more localized. This discrepancy suggests that the model learns distinct pathway-morphology association patterns across risk states. Additionally, the Focal adhesion pathway shows relatively clustered high-weight distributions in both case groups. The FoxO signaling pathway also exhibits variations in weight intensity and spatial distribution across cases, indicating that the model does not assign uniform attention patterns to all pathways but instead dynamically adjusts cross-modal weight allocation based on sample characteristics. The small window patches at the bottom of the figure correspond to high-weight regions in each pathway heat-map, providing local histomorphology information. This allows the process of “which regions the model focuses on under which pathway conditions” to be visualized intuitively. It is crucial to emphasize that attention weights reflect the correlation strengths learned by the model during multimodal fusion rather than the actual biological activation levels of molecular pathways. Overall, these visualizations reveal distinct spatial attention patterns for pathways across different risk phenotypes, demonstrating that MCAMamba can learn structured molecular-morphological correlation features, thereby providing interpretable support for risk stratification.
5. Discussion
Existing multimodal survival prediction methods still face two major challenges: efficiently capturing global dependencies in high-dimensional long-sequence features while maintaining linear complexity, and preserving informative modality-specific representations while enabling effective cross-modal interaction. To address these challenges, we propose MCAMamba, a multimodal method with bidirectional cross-attention and a state space model for cancer survival prediction. This method aims to extract both intrinsic intra-modal information and complementary inter-modal information from pathological images and genomic profiles, enabling efficient modeling and fusion of cross-modal features. Specifically, for the pathology modality, we employ PCL to cluster pathological images and select representative regions. Subsequently, by integrating the pathology encoder and decoder, the model captures long-range dependencies and spatial structural consistency within the sequence of pathology patches while maintaining computational efficiency. For the genomic modality, gene enrichment analysis is performed on genomic data for each cancer type using biological pathway information from the KEGG database. Subsequently, by combining the gene encoder and decoder to model pathway-level gene sequences, the model captures dependencies in complex molecular mechanisms and ensures biological interpretability. Building upon this foundation, we embed a Bidirectional Cross-Attention module within the encoder–decoder architecture. This framework explores intrinsic correlations between pathology and genomics, enabling bidirectional information exchange and complementary feature transfer across modalities. Finally, self-attention pooling aggregates global representations from all modalities, yielding more accurate survival predictions.
To validate the effectiveness of the MCAMamba, we conducted extensive experiments on four public TCGA cancer datasets. The results show that MCAMamba significantly outperforms multiple representative baseline models in survival prediction tasks, fully demonstrating its superior performance in multimodal cancer survival prediction. To further assess the contribution of key modules to predictive performance, we designed ablation experiments to systematically analyze the roles of patch clustering, the SR-Mamba encoder, the BCA module, and the SAP. However, the current results primarily demonstrate the validity of the model within the TCGA dataset. Independent external validation datasets have not yet been incorporated, and cross-cohort evaluation has not been performed. In addition, Kaplan–Meier survival curves were used to visualize the risk stratification results, which showed that MCAMamba has strong risk stratification capability. We also conducted an interpretability analysis of the model’s predictions from three perspectives based on representative high-risk and low-risk cases. However, this interpretability analysis remains preliminary and lacks systematic case-level evaluation. In future work, we will further assess the model’s clinical utility and generalizability through specific case studies and validation on independent external datasets.
Although the method proposed in this study has achieved significant results in multimodal cancer survival prediction, it currently relies primarily on pathological image representations at a single resolution. This limitation partially constrains the model’s ability to fully exploit the inherent pyramidal structure of WSIs, thereby affecting the accuracy of survival predictions. WSIs at different magnifications present hierarchical microscopic views, revealing multi-scale pathological information ranging from tissue phenotypes (5×) to cellular structures (20×) and even individual cells (40×). Based on this observation, future research will introduce multi-scale modeling strategies to comprehensively extract feature information at different scales and capture complementary morphological patterns, thereby enhancing cross-modal alignment and fusion capabilities between pathological and genomic features.
6. Conclusion
In this study, we propose MCAMamba, a multimodal cancer survival prediction method based on bidirectional cross-attention mechanisms and state space model. The proposed method is designed to efficiently model intra-modal representations of pathological images and genomic profiles while exploring cross-modal correlations and complementary information, thereby improving cancer survival prediction accuracy. Specifically, MCAMamba adopts a parallel encoder–decoder architecture to model pathological images and genomic data separately, extracting inherent valuable feature representations. Building on this architecture, we embed a Bidirectional Cross-Attention module to capture cross-modal correlations between the pathological and genomic modalities, while facilitating bidirectional information exchange and complementary feature transfer. Finally, Self-Attention Pooling aggregates global representations across modalities to achieve more accurate survival prediction. Experimental results on four public TCGA cancer datasets demonstrate that MCAMamba achieves significant improvements in survival prediction, with the c-index increasing by 2.47%- 17.9%.
Footnotes
Ethical considerations
This study constitutes a secondary analysis of publicly available data obtained from the open-access Cancer Genome Atlas (TCGA) repository. The TCGA project obtained ethics committee approval and informed consent from participants during the data collection phase. This research exclusively uses de-identified public data for analysis, involves no personally identifiable information, and requires no additional participant recruitment or intervention. Therefore, this study typically does not require further institutional ethics review.
Consent to participate
Informed consent was obtained from participants during the data collection phase of the original TCGA study. This study uses only de-identified public data for secondary analysis and does not require additional individual informed consent.
Author contributions
All authors contributed to this work. Writing—original draft, HY.C.; Writing—review and editing, WP. D, WX. W; Collect and process data, XF. L. All authors have read and agreed to the published version of the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data generated or analyzed during this study are included in this article and its supplementary materials.