
Abstract
Introduction
Gaucher’s disease is a lysosomal storage disorder caused by the deficiency of glucocerebrosidase. Gaucher’s disease has three clinical types: non-neuronopathic (Type 1), Acute Neuropathic (Type 2) and chronic neuronopathic (Type 3). The chronic neuronopathic (Type 3) is characterised by a variety of disease variants with onset in childhood with hepatomegaly, skeletal lesions and later slow horizontal saccades, treatment-resistant generalised tonic–clonic and myoclonic seizures, dementia, progressive spasticity, cognitive deterioration, ataxia and death in the second or third decade of life.
Case presentation
We describe a case of a 17-year-old girl who was born normally but subsequently developed treatment-refractory seizures at the age of nine with myoclonus, oculomotor apraxia, ataxia and cognitive decline. Enzyme activity of beta-glucocerebrosidase was found to be low without visceromegaly or bone involvement.
Conclusion
Screening for lysosomal enzyme activity should be done in patients exhibiting features suggestive of progressive myoclonic epilepsy.
Introduction
Gaucher’s disease (GD) was named after Philippe CE Gaucher who discovered it in a 32-year-old woman with splenomegaly and an abnormal cell named as Gaucher cell which is characterised by a crumpled silk appearance of the cytoplasm and eccentrically placed nucleus. The disease is caused by diminished activity of glucocerebroside cleaving enzyme (now known as enzyme acid b-glucosidase glucocerebrosidase (GlcCerase)).
GD is one of the commonest lysosomal storage diseases. It is a genetic disease in which glucosylceramide (GlcCer) accumulates in lysosomes of macrophage lineage. These GlcCer-laden macrophages are the classical hallmark of the disease and denoted as Gaucher cells. As GlcCer is an important constituent of biological membranes and is a key intermediate in the biosynthetic and degradative pathways of complex glycosphingolipids, its accumulation in GD is likely to have serious pathological results.1,2 Gaucher cells displace normal cells and accumulate in visceral organs, bone marrow and bones. This is a pan-ethnic disorder. Inheritance is recessive with a prevalence of 1 in 100,000. 3 In most countries, 5%–10% exhibit a neuronopathic phenotype. 4 More than 200 genotypes have been identified.
There is variability displayed in the severity of the disease ranging from asymptomatic to potentially lethal in the perinatal period. There are myriads of presenting features and it can present at birth or during juvenile, adolescent and adult onset. There are three clinical types of which Type 1, the non-neuronopathic type, is characterised by mild and slowly progressive disease which is underdiagnosed or a more severe type where there is severe visceromegaly and bone involvement. The manifestation also depends on the age of onset, with young onset disease being characterised by a more severe clinical course as compared to some adult onset cases. 5 Patients may have reduced quality of life with bone pain, osteonecrosis, fractures, skeletal growth retardation, marrow infiltration with visceral involvement such as hepatosplenomegaly, pulmonary problems (for instance, restrictive lung disease) as well as skin, eye and heart involvement. Some even have increased risk for neoplastic disease, delayed puberty and thymic involvement. 6
The neuropathic types are type 2 and type 3. In type 2, which can be acute, the disease is characterised by rapid onset within the first six months with rapid progression, typically early mortality, as the rule. These patients have systemic involvement with brainstem disease, spasticity, retroflexed head, strabismus, oculomotor apraxia, trismus, dysphagia and stridor. However in type 3, the disease comprises patients with early childhood onset who may have hepatomegaly, skeletal involvement, with early development of saccadic eye movement followed by seizures of myoclonic and generalised types which are treatment refractory. Patients also have pyramidal and cerebellar involvement with dementia, ataxia and regression of cognitive functioning. 8 Lately, a variant seen in adults with Parkinsonian features has been reported. 9 Some have reported a variant associated with cardiac valve calcifications, cloudy corneas, mental retardation and oculomotor ataxia.
Case report
We report a 17-year-old girl referred to the Neurology Clinic in 2008 at the age of 13 years for difficult-to-treat epilepsy. Her epilepsy had started at the age of nine years wherein she had generalised seizures starting with myoclonus. Birth and early developmental history was within normal limits. She initially attended normal school, although it was at this point she developed clumsiness and myoclonus, which progressed slowly over the next few years. By the age of 14, the myoclonus worsened with the onset of tremors, ataxia and cerebellar signs resulting in unsteady gait and progressive difficulty in walking and frequent falls. This was associated with deterioration and slowing of speech and progressive cognitive decline. Eventually, she stopped attending school at the end of form two (aged 14 years). Neurological examination showed an alert, small-built girl with generalised myoclonic jerks (spontaneous and stimulus-sensitive), oculomotor apraxia, cerebellar signs, intention tremor with spasticity and difficulty to ambulate, and difficulty talking due to dysarthria. She had no hepatosplenomegaly, nor hematological, skeletal, or respiratory involvement until now. With the disease progression, the patient started to have difficulties with oral intake and swallowing, and is currently on Ryle’s tube feeding, with associated weight loss - her weight is 40 kg (below the third centile). At present, she continues to have myoclonic jerks and occasional oculomotor apraxia, with the absence of neurobehavioural symptoms. The investigations carried out are summarised below.
Neurophysiology and neuroimaging
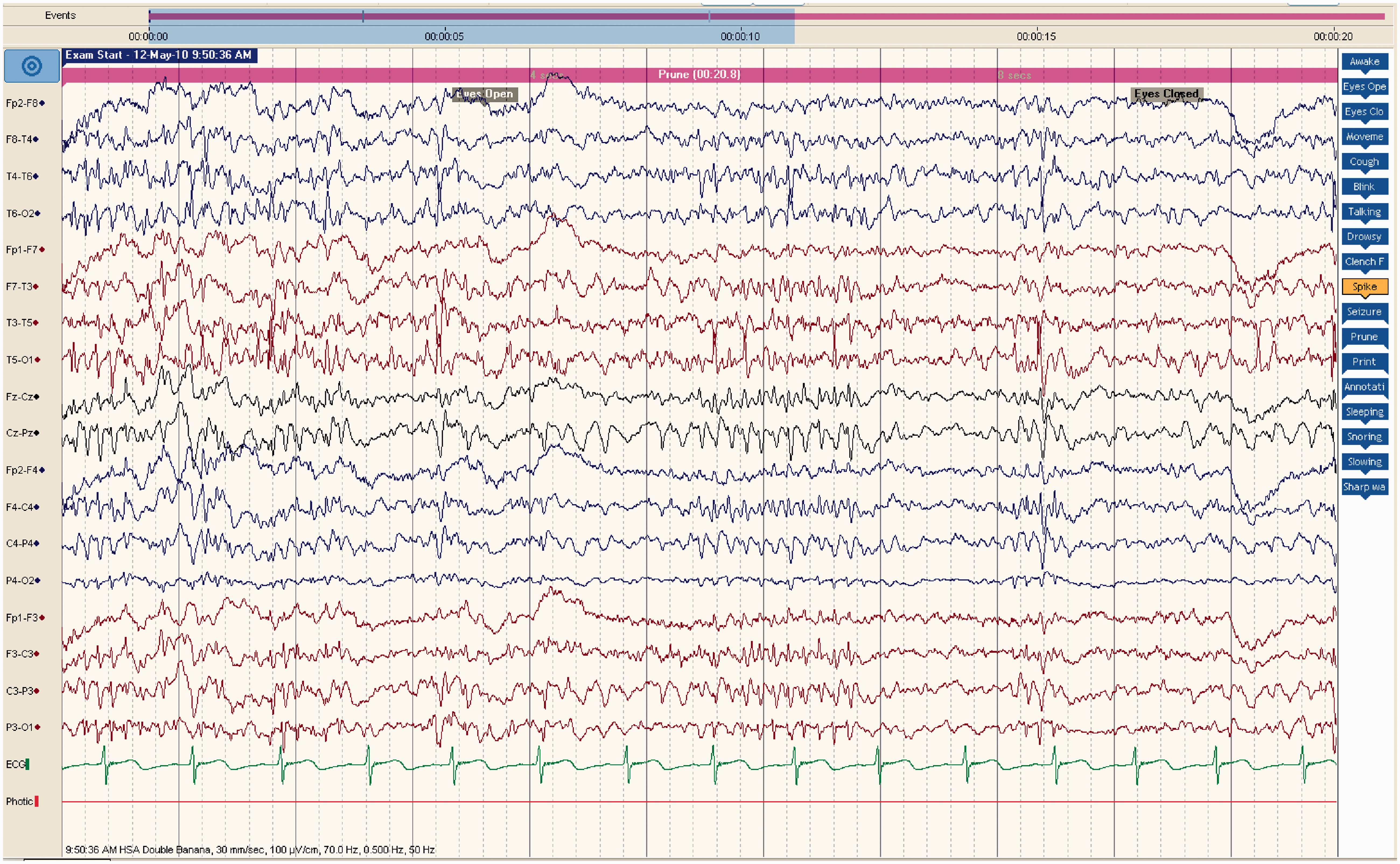
Electroencephalogram (EEG) done in May 2010 showed evidence of bifronto-temporal and central diffuse spikes and spike and slow waves (Figure 1). Computed tomography (CT) brain in April 2009, August 2010 and February 2011 showed no significant findings. Magnetic resonance imaging (MRI) brain in 2010 showed no structural abnormalities except for cerebral atrophy. EEG showing multifocal spikes, spikes and slow waves with runs of spikes occurring diffusely lasting between 1 and 1.5 s seen bifronto-temporally and centrally.
Laboratory results
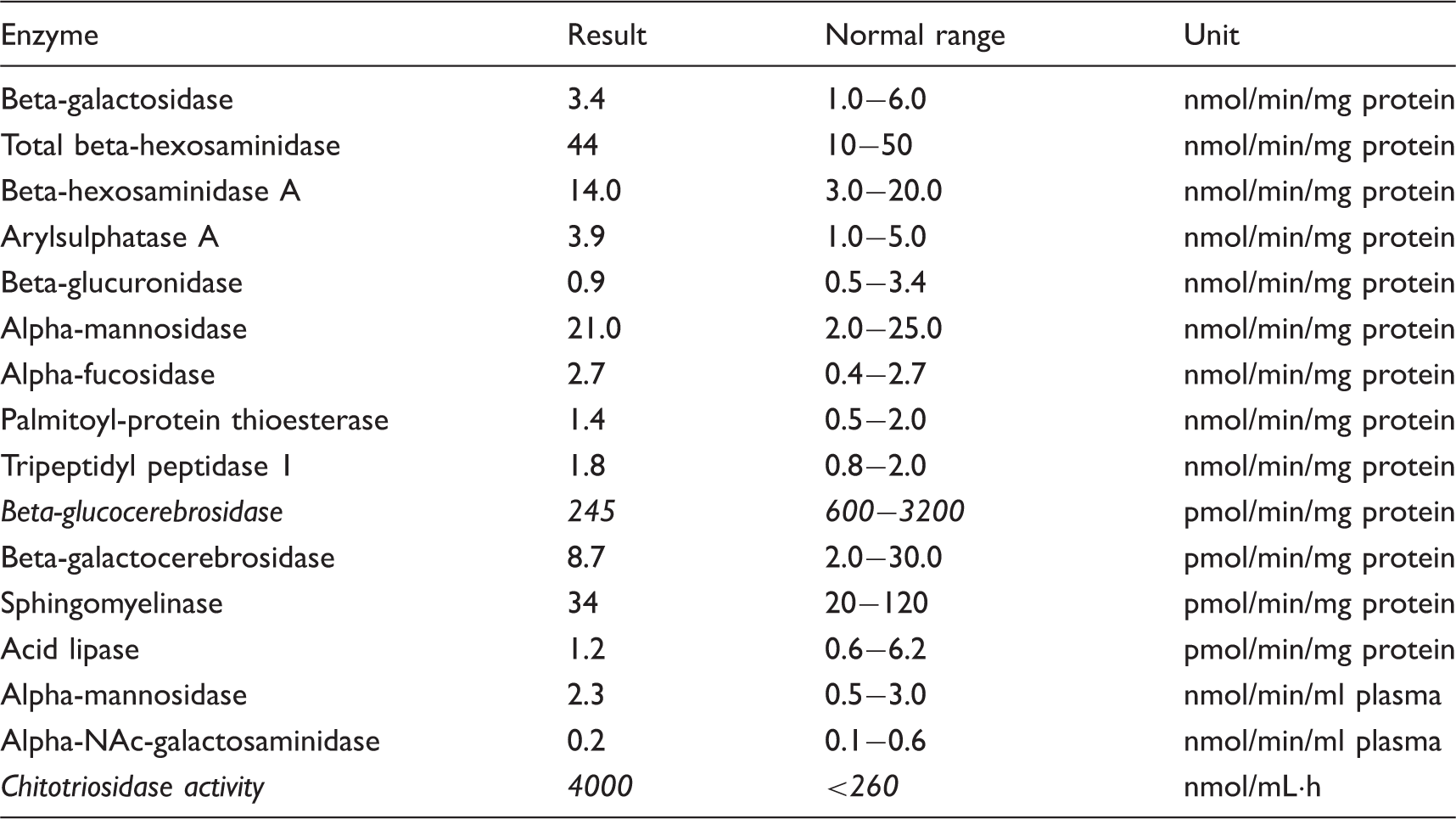
White cell lysosomal enzyme assay (Table 1). Marked decrease of the beta-glucocerebrosidase level (245 (normal 600–3200) pmol/min/mg protein) and an increase in the chitotriosidase activity (4000 (normal < 260) nmol/mL·h). Full blood count, full blood picture, renal profile, liver function test, calcium, creatinine kinase, serum ammonia, serum lactate, thyroid function test, serum copper, serum ceruloplasmin and serum B12 were all normal. Plasma amino acid: Non-diagnostic pattern. Urine organic acid: No abnormalities detected. Urine purine and pyrimidine analysis: Normal. Urine sialic acid: Normal. Urine oligosaccharide: No abnormalities detected. MERRF mutation analysis: No mutation found. Skin biopsy: No Lafora inclusion bodies seen. Genetic analysis was not done. White cell lysosomal enzyme assay.
This case is unique in that she has a progressive myoclonic epilepsy syndrome poorly responsive to antiepileptics with features of oculomotor apraxia, pyramidal and cerebellar signs with lysosomal enzyme assay confirming it to be neuronopathic GD Type 3a in the absence of severe visceral involvement.
Discussion
Progressive myoclonic epilepsies are made up of a group of disorders which include Unverricht–Lundborg disease, Myoclonic epilepsy and ragged red fibers (MERFF), Lafora disease, neuronal ceroid lipofuscinoses and Type 1 sialidosis as well as less commoner causes such as atypical inclusion body disease, dentatorubropallidoysian atrophy (DRPLA), non-infantile neuronopathic form of Gaucher disease and Niemann–Pick type C.
Occasionally, early on, this group of diseases may be difficult to differentiate from idiopathic generalised epileptic disorders. However, a high index of suspicion should be there if patients develop psychomotor retardation and cognitive deterioration, refractory seizures unresponsive to treatment with associated dysmorphic features, skeletal abnormalities, visceromegaly and other associated movement disorders such as tremors, choreas, ataxias and eye movement defects associated with multiple diffuse and independent spike wave complexes with underlying encephalopathy features on electroencephalogram. Differentiating between all these disorders requires pattern recognition, careful history taking and neurological examination, and careful fundic examination, for example, the cherry red spot in Niemann–Pick type C coupled with ancillary paraclinical testing. Among the tests which would be useful would be neurophysiologic testing, i.e. looking for specific patterns on electroencephalograms and electroretinograms, nerve conduction studies looking for evidence of myopathy, blood tests including blood and cerebrospinal fluid for lactate and lysosomal and genetic studies.
In this case, we highlight a rare case of Type 3 neuronopathic GD who presented initially just like a case of juvenile myoclonic epilepsy but later on developed progressive cognitive deterioration, worsening refractory epilepsy, ataxia, oculomotor problems and tremors becoming more dependent with time in the absence of visceromegaly clinically with associated myoclonic episodes. Extensive investigations were done looking for the other differential diagnosis as mentioned above such as MERFF, but serum lactate and MERFF mutation analyses were negative, skin biopsy did not show any Lafora bodies or any other inclusions and urine sialic acids were within normal limits. MRI of the brain showed mild cerebral atrophy but there were no changes of DRPLA or any other changes suggestive of other progressive myoclonic epileptic disorders. After careful consideration of the phenotype, Type 3 non-neuronopathic GD was the final consideration.
Type 3 GD has highly variable manifestations in the central nervous system (CNS) and viscera. It can present in early childhood with rapidly progressive visceral disease and slowly progresses to static CNS involvement or in adolescence with dementia or in early adulthood with mild visceral disease and refractory progressive myoclonic seizures as we see in our case. Sometimes, neurological findings may only be defects in lateral gaze tracking, which may remain static for decades. Mental retardation is another neurological problem, which can be slowly progressive or static. The visceral, but not the CNS, involvement responds to enzyme therapy. 10 Type 3 GD is further sub-classified into three subtypes: Type 3a is a progressive neuronopathic disease with onset in adolescence (that includes progressive ataxia, myoclonus and dementia as in our case); 11 Type 3b has extensive hepatosplenomegaly and bone disease but non-progressive oculomotor apraxia, and Type 3c (cardiovascular form) is rare and characterised by oculomotor apraxia, corneal opacities and cardiovascular calcification, with little visceral disease. 11
Diagnosis is usually made by enzyme analysis showing reduced activity of glucocerebrosidase activity in peripheral leukocytes. 12 It is a valid and easy method to diagnose GD. 13 In many cases, the disorder is diagnosed by bone marrow examination (which was not done in our case). Other blood tests would include measuring the activity of serum acid phosphatase and angiotensin-converting enzyme, lysosomal enzymes activity, DNA mutational analysis, biopsy of the spleen, MRI, CT and X-ray of the skeleton. Biochemical markers such as chitotriosidase have been identified as well in this disease and they tend to rise when patients become symptomatic. Prenatal diagnosis can be made through sampling from the chorionic villus or through amniocentesis. 14 Management of these conditions is usually supportive. To date, there are more than 300 mutant alleles identified. For example, the presence of at least one N370S allele is associated with Type 1 disease whereas homozygosity for L444P generally predicts Type 3 disease. 10 Enzyme replacement therapy is available but this is more effective in GD patients with visceral but not CNS involvement. However, enzyme replacement treatment with intravenous recombinant glucocerebrosidase is the standard treatment for Types 1 and 3 for more than 15 years. 15 It is noted that the level of chitotriosidase is not as high as we would routinely see in patients affected by GD. However, this might be due to the lack of visceromegaly.
Conclusion
This patient exhibited a progressive debilitating disorder characterised by refractory generalised seizures with myoclonus, poor response to treatment with oculomotor apraxia and cerebellar features. Her diagnosis was delayed till the age of 17 years as lysosomal enzyme assays were not routinely done and due to the low index of suspicion more so in the absence of organomegaly and skeletal involvement which may develop later on during follow-up.
Footnotes
Acknowledgements
The authors would like to thank the Director of Health Malaysia for granting permission to publish this paper. They would also like to express their thanks to the Director of Hospital Sultanah Aminah Johor Bahru, Malaysia as well as the Head of the Medical Department and the Neurophysiology team.
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.