
Abstract
Background
Immune-mediated necrotising myopathies are characterised clinically by the subacute onset of proximal limb weakness, accompanied by elevated creatinine kinase levels. They are distinguished from other myopathies by the absence of prominent infiltration of the muscle with inflammatory cells in the biopsies.
Case presentation
A 44-year-old man presented with upper extremity weakness and dysphagia. Laboratory tests included a creatinine kinase level of 4362 U/L (normal: 52–336 U/L). Rheumatological markers were all negative. A muscle biopsy showed multiple necrotic fibres with minimal inflammatory infiltration. One gram of methylprednisolone (IV) was given, followed by 1 mg/kg of methylprednisolone daily by the oral route. Intravenous immunoglobulin (0.4 mg/kg/day) was given for five days. Muscle weakness regressed and dysphagia disappeared with treatment. The patient remains well in the 23rd month of treatment, taking 5 mg/day prednisolone and monthly intravenous immunoglobulin.
Conclusion
Treatment of immune-mediated necrotising myopathy can be challenging as evidence-based therapeutic options are limited. It is generally accepted that early and extensive immunosuppression, including glucocorticoids as first-line agents, may be required.
Introduction
Immune-mediated necrotising myopathies are recognised as a subgroup of idiopathic inflammatory myopathies. 1 They are characterised clinically by the subacute onset of proximal limb weakness, elevated creatinine kinase (CK) levels and myopathic findings on electromyography. They are distinguished from other inflammatory myopathies by the presence of necrosis and the absence of prominent infiltration in muscle biopsy. 2 Recognition is important because of their potential to respond to immunosuppressive treatment.
Herein we describe a patient with necrotising myopathy which showed a good response to immunosuppressive therapy, highlighting the importance of early recognition.
Case presentation
A 44-old man presented to our clinic with upper extremity weakness and dysphagia with solid nutrients. He also complained of weight loss of about 30 kg in the past three months. He had fatigue during daily routine activities. A week previously, a CK level of 4267 U/L had been measured in another hospital. His medical history was insignificant other than a smoking history of 20 pack-years. He had no rash, arthralgia, fever or respiratory symptoms. He denied any regular medication or toxin exposure, including statins, alcohol, herbal medicines and illicit drugs.
Physical examination showed bilateral proximal weakness in arms (Medical Research Council grade 2/5) and legs (grade 3/5). There was also a spotty-red pigmentation over the patient’s right shoulder which was considered as non-specific (Figure 1).
Laboratory tests
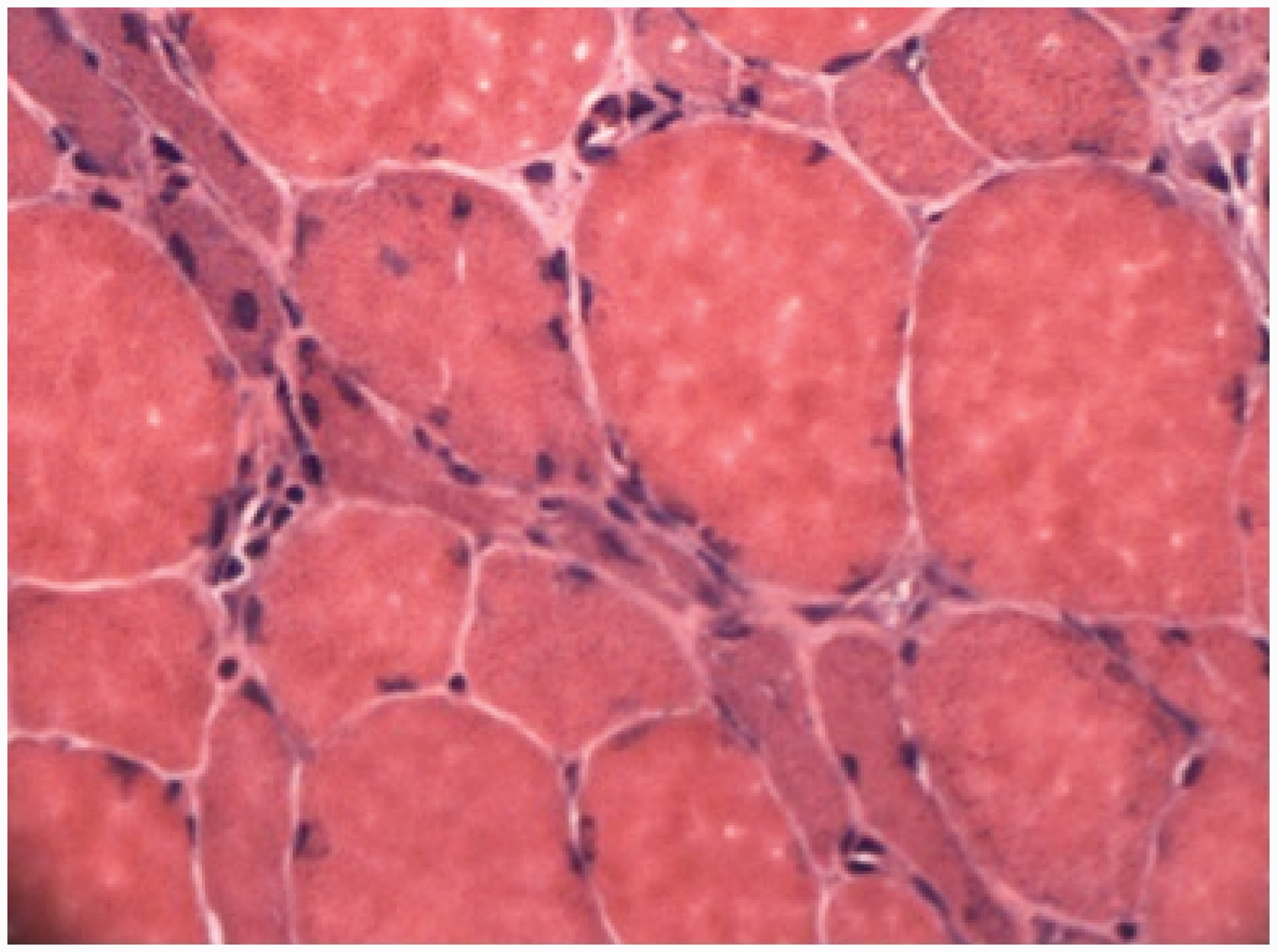
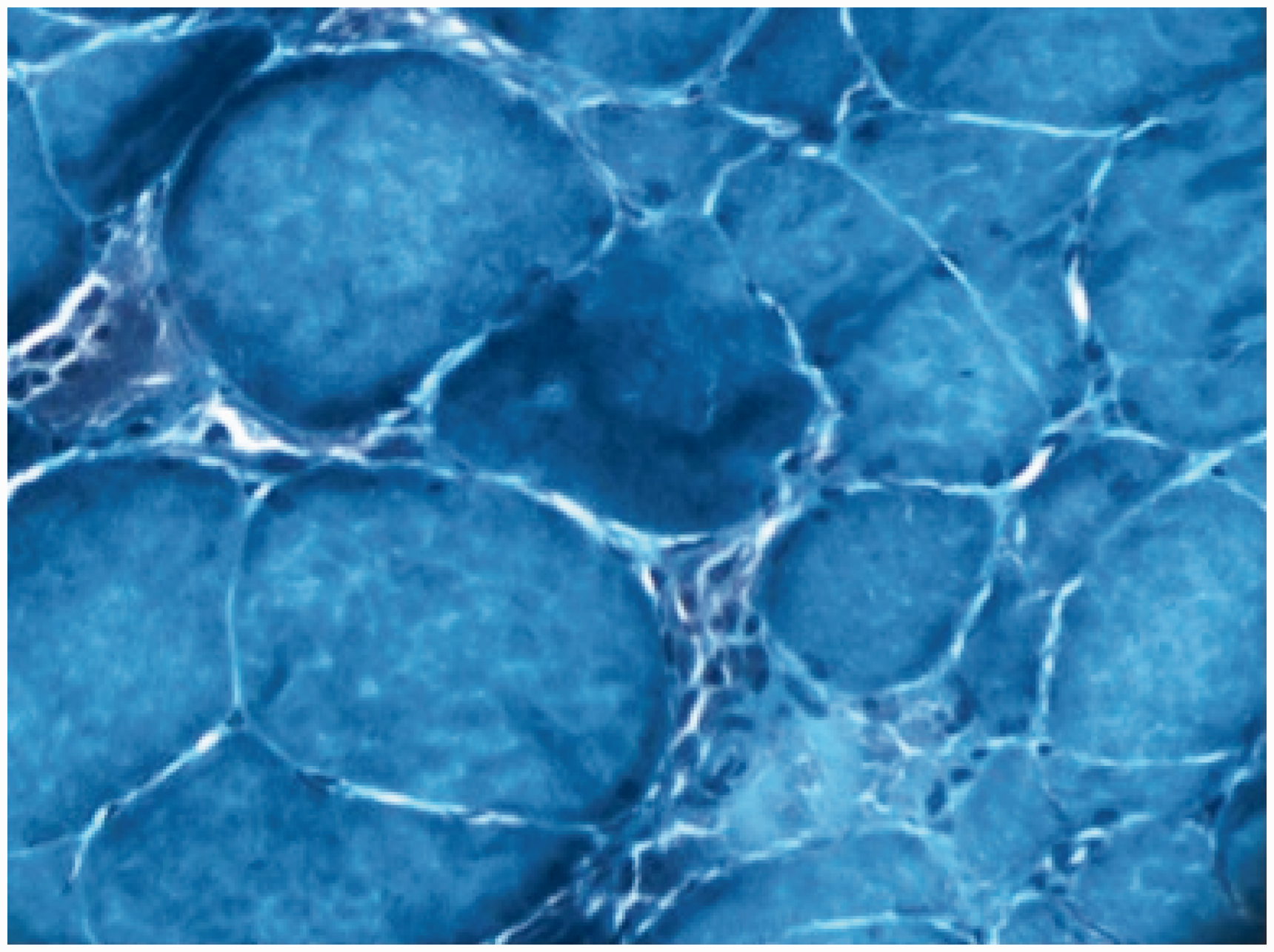
CK level was 4362 U/L (N < 171). Acute phase reactants were within normal limits. Thyroid-stimulating hormone level was 0.84 mU/L (0.34–5.6). Viral serologies were negative. Rheumatological markers were all negative including Anti-nuclear antibody (ANA), Extractable nuclear antigen (ENA) and rheumatoid factor. Myositis-specific autoantibodies including anti-Jo1 and anti-SRP antibodies were negative. Measurement of Hydroxy methylglutaryl coenzyme-A (HMG-CoA) reductase was not available. Capillaroscopy examination was normal. After electromyography showed a myopathic pattern, a muscle biopsy was performed to delineate underlying pathology. This showed multiple necrotic fibres without inflammatory infiltration and without an increase of endomysial connective tissue (Figures 2 and 3). A diagnosis of immune-mediated necrotising myopathy was made.
Volumes of right deltoid muscles (a) before and (b) after the treatment. Biopsy material showing multiple necrotic fibres with minimal inflammatory infiltration (H&E stain.). Biopsy material showing multiple necrotic fibres with minimal inflammatory infiltration (Gomori trichrome stain.).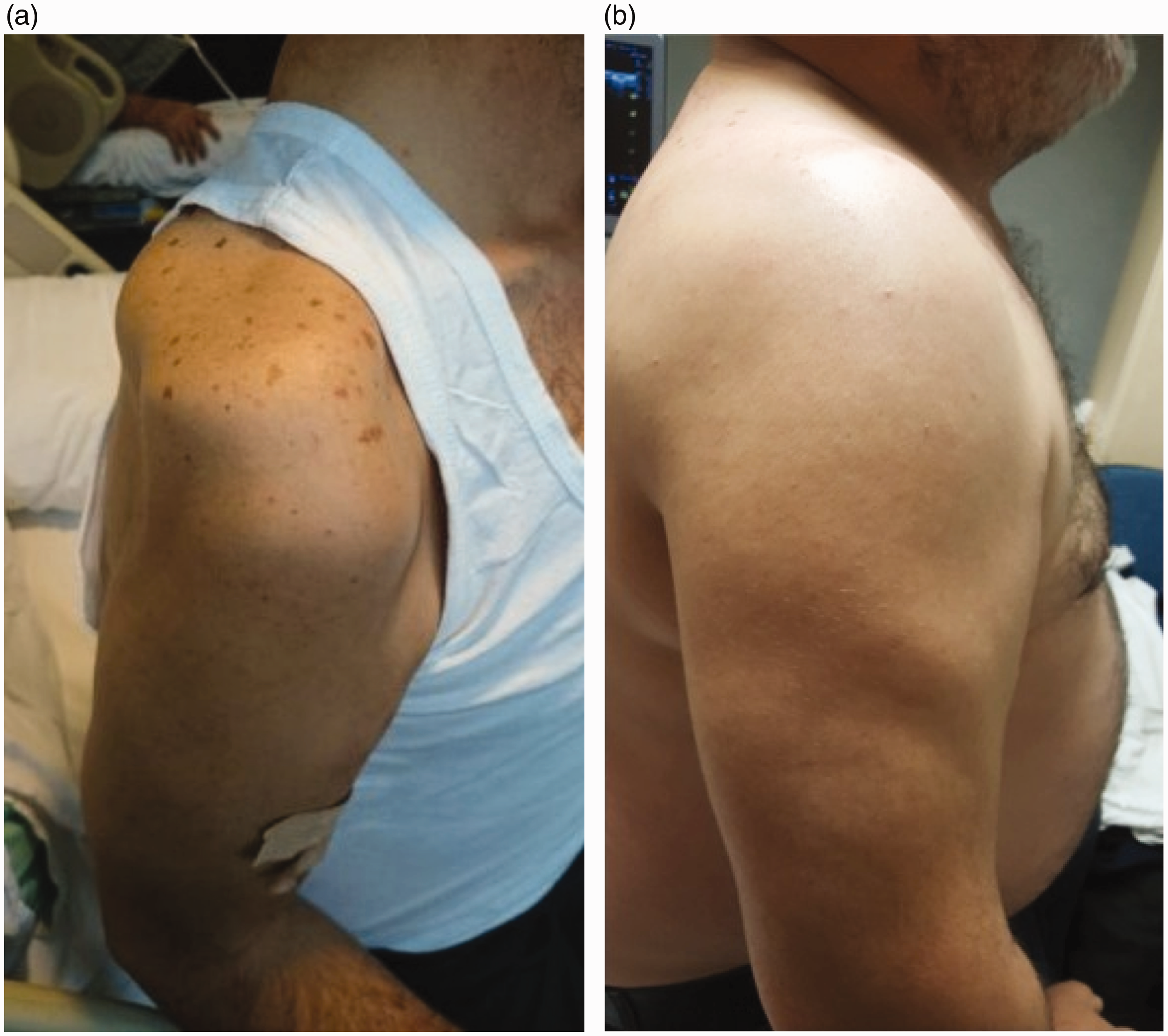
A single dose of 1 g methylprednisolone IV was given to the patient, followed by 1 mg/kg of methylprednisolone IV daily. An extensive search for an occult malignancy was made. Chest and abdomen computed tomography scans were normal except for a few non-specific ground glass opacities. Upper and lower gastrointestinal endoscopies revealed no signs of malignancy. Positron emission tomography (PET) scan showed a mucosal mass in the piriform sinus. Nasopharynx MRI was performed and confirmed the mass in piriform sinus with extensions to aryepiglottic fold and left paraglottic space. A biopsy under direct laryngoscopy revealed a squamous papilloma, which is a benign proliferative lesion. Although CK levels started to drop in two days, 0.4 g/kg/day of intravenous immunoglobulin (IVIG) was given for five days because of increased dysphagia and weakness. Muscle weakness regressed, and dysphagia disappeared within a few days of immunoglobulin treatment. CK levels dropped to 1843 U/L within four days. The patient was discharged, taking 60 mg/day prednisolone with a ‘steroid taper’ scheme to begin after four weeks and with the plan of monthly IVIG. Two months after discharge, muscle strength in upper and lower extremities was increased to 4/5. IVIG treatment was continued monthly. Muscle enzymes became elevated at the 12th month of treatment, but owing to stable muscle strength, no change was made in the treatment regimen (Figure 4). The patient remains well in the 23rd month of treatment, taking 5 mg daily of prednisolone.
The course of CK and LDH levels during treatment. Blue line accounts for CK levels and red line accounts for LDH levels.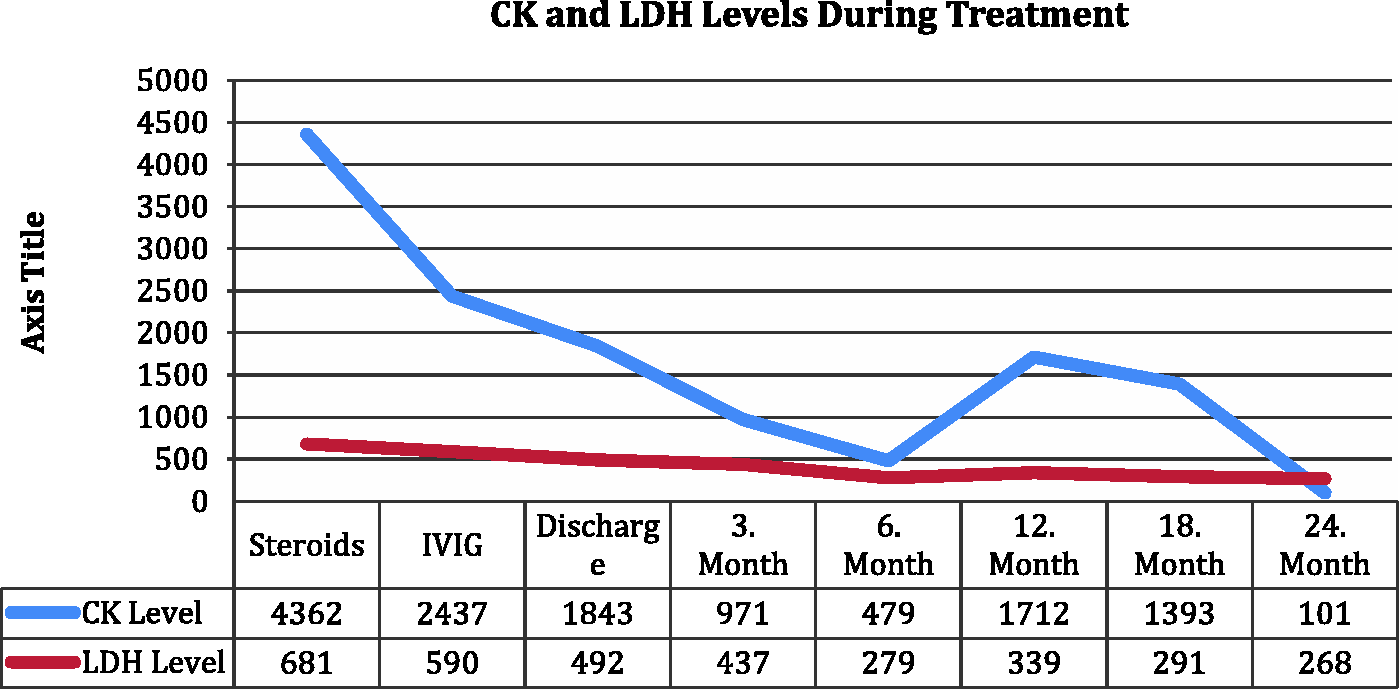
Discussion
Necrotising myopathies without inflammation have been described for decades but were reported as polymyositis (PM) or dermatomyositis (DM). 3 By the early 1990s, a group of necrotising myopathies had been histologically distinguished from these entities. Increasing recognition of this syndrome led to the proposal for a separate classification for necrotising autoimmune myopathy (NAM) by Amato in 2004. 4
The annual incidence of inflammatory myopathies is about two to seven per million, and NAM, which is considered a rare disease, accounts for around 20% of inflammatory myopathies. 5
In the 119th European Neuromuscular Centre Workshop in 2003, NAM was classified as a subgroup of inflammatory myopathies by fulfilling clinical inclusion criteria of adult DM or PM, such as onset over 18 years, subacute or insidious onset of symmetrical proximal > distal limb weakness and neck flexor > neck extensor weakness, elevated serum CK levels and detection of myositis-specific antibodies, as well as exclusion criteria such as clinical features of sporadic inclusion body myositis, ocular weakness, isolated dysarthria and signs of toxic, endocrine myopathy, family history of muscular dystrophy or proximal motor neuropathies (e.g. spinal muscular atrophy). 6
The diagnosis of NAM relies on the muscle biopsy findings in the appropriate clinical context. These are distinguished by marked necrosis of muscle fibres with minimal or absent inflammation and with macrophages being the main effector cells in contrast to CD8 cells in PM. In muscle biopsies, macrophages seen mostly around the necrotic fibres and sparsely distributed in the endomysium. 7
NAM can be divided into subgroups, according to aetiology, as anti-SRP related, connective tissue disease-related, statin-triggered, viral and paraneoplastic. 8
While presentation is similar to PM and DM with a subacute onset of symmetric limb weakness, the onset of NAM can be more acute and severe – perhaps occurring over a few days. Other symptoms include fatigue, weight loss and myalgia. The most severely affected muscles are usually the deltoid and psoas. Pharyngeal and respiratory muscles may also be involved, causing dysphagia and dyspnea. 9 CK is often highly elevated, usually greater than 10 × upper limit of normal, but cases with lower CK levels have been reported.10,11 Capillaroscopy changes – including microhaemorrhages, capillary enlargement and ramified capillaries – are seen frequently in PM and DM patients, but the role of capillaroscopy is yet been to be defined in NAM. 12 Some muscle-specific autoantibodies have been associated with NAM including anti-SRP and anti-HMG-CoA reductase, which should be checked whenever testing is available. However, seronegative cases are common, especially in malignancy-associated cases. 13 In patients with anti-SRP antibodies, severe limb muscle weakness, neck weakness, dysphagia, respiratory insufficiency and muscle atrophy are more frequent.14,15 Owing to its association with malignancy, the finding of necrotising myopathy should prompt a search for underlying disease. 16
In view of the disease’s recent recognition, and the absence of randomised studies, treatment can be challenging. However, it is generally accepted that NAM requires early and extensive immunosuppressive management, including glucocorticoids. 5 The most used steroid-sparing agents are azathioprine, methotrexate and cyclosporine. 17 Cyclophosphamide may be an option for severe disease or interstitial lung disease secondary to NAM. 18 A recent report from the Mayo Clinic suggested that early treatment with IVIG associated with a significantly better clinical response, defined as an increased likelihood to attain marked improvement or return to baseline at six months, but this difference was not sustained in the follow-up. 16 Rituximab can be useful in patients with anti-SRP antibodies. 19 Our patient showed a very good response to the combination of steroids and IVIG. Although muscle enzymes were dropping, muscle weakness did not improve until the beginning of IVIG treatment.
Conclusion
Immune necrotising myopathy is a unique form of inflammatory myopathy with distinct clinical and pathological features. Although good quality evidence is lacking, early treatment with steroids and steroid-sparing agents including immunoglobulins may improve outcomes in this patient group.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.