
Abstract
Magnetic nanoparticles (MNPs)/polymer composite nanofibers were prepared via electrospinning of polyacrylic acid (PAA)/polyvinyl alcohol (PVA) aqueous solutions with homogenously dispersed magnetite Fe3O4 nanoparticles (NPs). Experiment results showed that the diameters of Fe3O4 NPs were in a range of 10 nm to 40 nm under all of our synthesis conditions; and Fe3O4 NPs can homogenously disperse in PVA composite nanofibers when they were stabilized by PAA; the crystallinity of PAA/PVA nanofibers increased along with the increase in Fe3O4 NPs content. The magnetic property of Fe3O4 NPs/PAA/PVA composite nanofibers was also investigated and the results showed that the composite nanofibers show a soft ferromagnetic behavior which is the same as Fe3O4 at room temperature. In addition, the water resistance of Fe3O4 NPs/PAA/PVA composite nanofibers was dramatically enhanced by heat treatment. Therefore, in our process to preparing magnetic Fe3O4 NPs/PVA composite nanofibers, PAA acts not only as a dispersant but also as a cross-linking agent.
Introduction
Polymer magnetic nanocomposite systems have many potential applications in various fields. Thus, interest in the design and controlled preparation of these composite materials with magnetic property continues to increase.1−4 One of the most useful magnetic materials may be magnetic fiber, which is used for applications such as magnetic paper, health-care cloth, magnetic filters, electromagnetic wave adsorbents, and various other applications because of their high forming abilities.5−7 Traditionally, magnetic fibers have been produced by coating magnetite nanoparticles (MNPs) on the surface of the fibers.8−10 However, the magnetism of these products is not high enough for magnetic application because there may be not enough amounts of MNPs coated on the surface. Furthermore, the magnetism would weaken as a result of the losses of MNPs after post-treatments. To obtain a magnetic fiber with higher magnetism and higher stability, some researchers tried to incorporate the MNPs into a polymeric fiber matrix directly.11−14 Nevertheless, one of the main challenges is the ability to deliver well-dispersed nanoparticles with the desired composition as well as the prevention of aggregation.15−17 Surfactants can be used to resolve the issue and prepare homogeneous MNPs/polymer composite system, but their use will unavoidably sacrifice part of the mechanical property of the composite fibers.
Electrospinning is a relatively simple and inexpensive method to produce fibers with a diameter ranging from a few micrometers to a few nanometers.18−21 Due to their large surface areas and small sizes in comparison with conventional textiles, electrospun nonwoven mats have become excellent candidates for applications in filtration, biomedical films, and scaffolds for tissue engineering.22−25 This novel fiber-spinning technique provides the capacity to produce ultrafine magnetic composite fibers using a mixture of various polymers and magnetic nanoparticles.26−30
Polyvinyl alcohol (PVA) is one of the most popular polymers used for electrospun composite fiber production due to its thermostability, excellent biocompatibility and high mechanical performance.31−35 However, its applications are limited by its high hydrophilicity, through which it dissolves immediately on contact with water. Therefore, PVA nanofibers have usually been modified by either chemical or physical cross-linking to improve their water resistance and mechanical properties. 36
This paper describes the fabrication of MNPs/PAA/PVA composite nanofibers with homogeneous dispersion of Fe3O4 NPs and high water resistance. In our experiments, polyacrylic acid (PAA) was used as a polymer surfactant37,38 to stabilize the aqueous suspension of Fe3O4 NPs and facilitate their dispersal in PVA solutions, due to PAA being able to provide electrostatic and steric repulsion against particle aggregation. Furthermore, PAA was also used as a cross-linking agent to enhance the water resistance of the magnetic composite nanofibers through heat treatment. 39 This method provides a simple and convenient process to prepare a homogenous NPs/PVA composite system with high stability and can greatly improve the practical application of NPs/PVA composite.
Experimental
Materials
PVA HC with a degree of hydrolysis (DH) = 99.9 mol% and degree of polymerization (DP, is usually defined as the number of monomeric units in a macromolecule or polymer) = 1700 was kindly provided by Kuraray Co. Ltd, Tokyo, Japan. PAA (Mw = 5000), iron (II) sulfate heptahydrate (FeSO4·7H2O), iron (III) chloride anhydrous (FeCl3), ethanol, HCl solution and ammonia solution (28% NH3 in water) were purchased from Wako Pure Chemical Industries, Ltd., Japan. All chemicals were of chemical grade and were used without further purification. Distilled water was used as the solvent.
Preparation of magnetic Fe3O4 NPs
Magnetic Fe3O4 NPs were prepared by coprecipitating Fe2+ and Fe3+ ions in aqueous ammonia solution. 40 Next, 200 mL of a mixture of FeCl3 (0.01 mol/L) and FeSO4 (0.006 mol/L) at pH 2 was prepared under N2. Then, NH4OH aqueous solution (5 mol/L) was dropped into it with violent stirring until the pH of the solution rose to 10. The resulting precipitate was stirred for 30 min. After triple washes with deionized water and ethanol, the precipitates were re-suspended in 100 mL deionized water. The pure magnetic NPs were isolated from the solution by a magnet bar and dried in a vacuum oven at 40°C for 48 h.
Preparation of Fe3O4 NPs/PVA/and Fe3O4 NPs/PAA/PVA suspension
1.5 g Fe3O4 NPs were dispersed in 50 mL distilled water and 50 mL (5 wt%) PAA aqueous solution respectively, then both suspensions were treated by ultrasonic mixing for 30 min to get two kinds of 3 wt% Fe3O4 NPs suspension for reserve. PVA chips were dissolved in distilled water in a static rotary mixer (500 rpm) at 25°C for 1 h and then rapidly heated to 95°C and this temperature was maintained with high-speed stirring (1000 rpm) for 2 h to obtain a 30 wt% PVA solution. Varying amounts of 3 wt% Fe3O4 NPs suspension, PAA solution and water were then added to the as-prepared PVA solution to adjust the Fe3O4 NPs concentration in the PVA to 5, 10, and 15 wt% (weight of solid PVA content) while maintaining the PVA concentration at 8 wt% (PVA/PAA = 2:1) in each composite suspension with high-speed stirring. Finally, two kinds of suspension were obtained: PVA/Fe3O4 NPs and PVA/PAA/Fe3O4 NPs.
Electrospinning
The electrospinning setup (Kato Tech, Kyoto, Japan) used in this study consists of a syringe with a flat-end metal needle (1.20 mm× 38 mm), a syringe pump for controlling the feeding rate, a grounded cylindrical stainless steel mandrel, and a high-voltage direct current (DC) power supply. For the electrospinning procedure, the suspensions were loaded into a syringe coupled with nozzles and electrospun under a high DC voltage of 10 kV, with a distance from the needle tip to the collector of 15 cm and a feeding speed of 0.5 mL/h.
Annealing treatment and water resistance test
Immediately after spinning, the electrospun fibers were dried at room temperature in a vacuum oven for 24 h, and then the temperature was raised and maintained at 100°C for 2 h. The samples were allowed another 2 h to cool down before being removed from the oven. The nanofibers were immersed in 25°C water for several days separately. The samples were then transferred into isopropyl alcohol for 10 min before loading in a chamber to complete the drying process under vacuum (0.002 MPa) for later SEM characterization.
Characterization
Our samples were imaged with a field emission scanning electron microscope (FE-SEM, Hitachi S-5000, and 10 kV, sputtered with palladium-platinum), scanning electron microscope (SEM, Hitachi S-3000N, and 20 kV, sputtered with palladium-platinum) and a transmission electron microscope (TEM, JEM-1010, and 10 kV). Thermo-gravimetrical analysis (TGA) was performed on a Rigaku Thermo Plus TG8120 apparatus (Rigaku Denki, Japan) under air atmosphere at 5°C/min in a temperature range of 30°C to 850°C. The magnetic NPs and composite nanofibers were examined with an X-ray diffraction (XRD) pattern obtained using a Rigaku RINT-2550 at a voltage of 40 kV and a current of 40 mA with CuKα radiation (λ = 1.5406 Å), in the 2θ range from 3° to 90° at a scanning step of 2°/min. Magnetic measurements were performed on a commercial superconducting quantum interference device (SQUID) magnetometer (Quantum Design, MPMS). Hysteresis loops were measured at 300 K in a magnetic field in the range of −10 kOe to 10 kOe. The samples were also investigated by the Fourier transform infrared (FT-IR) spectroscopy before and after annealing treatment.
Results and discussion
Characterization of magnetic Fe3O4 NPs
Figure 1 illustrates the FE-SEM images (a) and TEM images (b) of the magnetic NPs, and we can see that the diameters of nanoparticles are monodispersed in the range of 10 nm to 20 nm. More detailed structural information of the products was provided by the high-resolution TEM (HRTEM) analysis, which shows the HRTEM image taken from an individual NP structure. Clear lattice fringes are observed in HRTEM image, indicating that crystalline particles formed in the hydrothermal process. Moreover, the measured spacing of the crystallographic planes is about 0.483 nm, which is close to that of the (111) lattice planes of magnetite crystals, and 0.298 nm which is close to that of (220) lattice planes of magnetite crystals.41,42
FE-SEM and TEM image of Fe3O4 NPs synthesized by coprecipitation method. (inserts are high-resolution images).
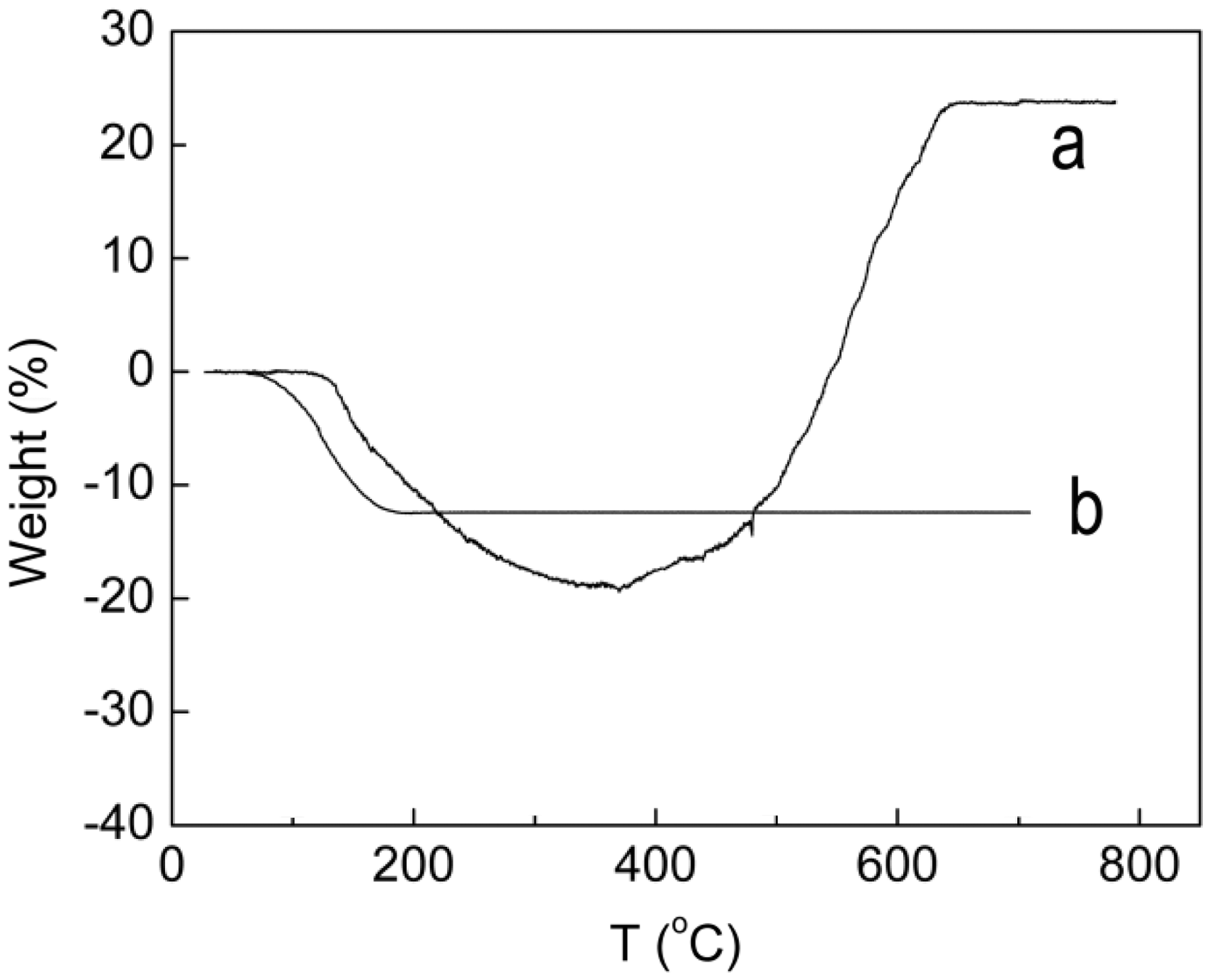
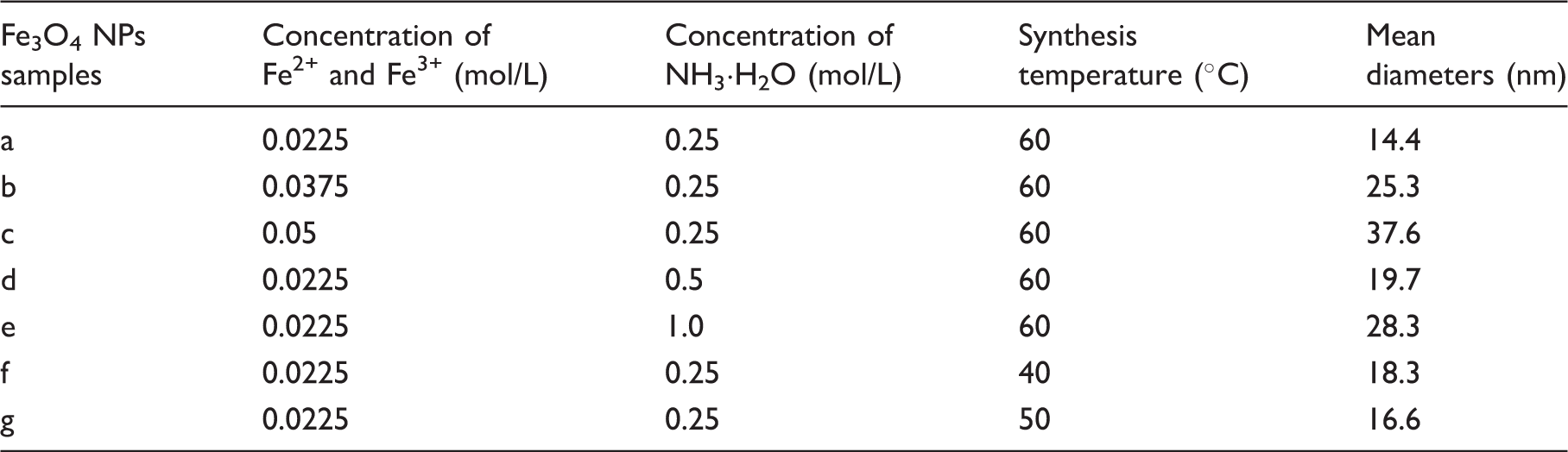
XRD was used to further characterize the magnetic NPs. As shown in Figure 2, the XRD patterns of the as-obtained magnetic NPs are in good agreement with those of the pure Fe3O4 (Magnetite, JCPDS No. 64-8075). In addition, the weight of magnetic NPs samples increased and their colors were changed form black to red during the TGA, which further proved the synthesized magnetic NPs were Fe3O4 (Figure 3). The intensity of the XRD pattern of the magnetic NPs also clearly increased along with the increase in synthesis temperatures, indicating that temperature has a positive influence on Fe3O4 NPs crystallinity. The effect of synthesis temperature, ferric ion concentration and ammonia solution on the NPs diameters was investigated and the results are shown in Table 1. The diameters of synthesized Fe3O4 NPs under all of our experiment conditions is no bigger than 40 nm and sample (a) with the smallest diameter was chosen as a filler for preparation of magnetic composite nanofibers.
XRD patterns of magnetic NPs synthesized at different temperatures. TGA of ferric oxide NPs (a. Fe3O4; b. Fe2O3). The diameters of synthesized Fe3O4 NPs under all of our experiment conditions
Characterization of magnetic Fe3O4 NPs/PAA/PVA composite nanofibers
The preparation of magnetic nanoparticles/polymer composite systems has been intensively pursued because of these systems’ broad applications, including magnetic storage media, ferrofluids, magnetic resonance imaging, and magnetically guided drug delivery. However, one of the main challenges facing almost all of these applications is the ability of the composite systems to deliver well-dispersed nanoparticles with the desired composition, structure, and uniformity as well as the prevention of aggregation. The principal cause of aggregation is the short-range forces (van der Waals attraction) between the two particles. 43 To counteract these attractive interactions and promote stability, equally short-range repulsive forces are required. These are enforced either by electrostatic repulsion between the particles or by coating the particles with organic long-chain molecules.
For fully hydrolyzed PVA aqueous solution, the viscosity is very high even in low concentration (<10 wt%), therefore, it is extremely difficult to prepare homogeneously dispersed NPs/PVA composite materials. In our experiments, PAA was used as a polymer surfactant to stabilize the aqueous suspension of NPs, which could provide electrostatic and steric repulsion against particle aggregation. As seen in Figure 4a and b, Fe3O4 NPs randomly formed irregular aggregation in PVA electrospun nanofibers (diameter is about 250 nm), which will cause heterogeneity of magnetism distribution in the magnetic nanofibers mat. But Fe3O4 NPs, which were formed prior to dispersal in the PAA solution, can homogenously disperse in PVA composite nanofibers (Figure 4c and d).
TEM images of Fe3O4 NPs/polymer composite nanofibers. (a and b: Fe3O4 NPs/PVA composite nanofibers; c and d: Fe3O4 NPs/PAA/PVA composite nanofibers).
The XRD pattern of Fe3O4 NPs/PAA/PVA composite nanofibers revealed the characteristic peaks of both the Fe3O4 and the PVA as shown in Figure 5. A wide peak was observed at 19.4°, which is in agreement with the XRD peak of the crystalline pure PVA nanofibers, while the other peaks are identical to Fe3O4 NPs. The intensity of the peak at 19.4° became stronger along with the content of Fe3O4 NPs, which indicated that the existence of Fe3O4 NPs facilitated the crystallization of polymer.
XRD patterns for the Fe3O4/polymer nanofibers mat.
The TGA curve for PAA/PVA nanofibers and Fe3O4 NPs/PAA/PVA nanofibers are shown in Figure 6. The figure shows the weight loss before 200°C resulted from the evaporation of bonded water in PAA/PVA.
TGA of PAA/PVA nanofibers and Fe3O4 NPs/PAA/PVA nanofibers.
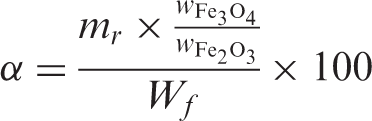
Content of Fe3O4 in magnetic nanofibers and the saturation magnetization
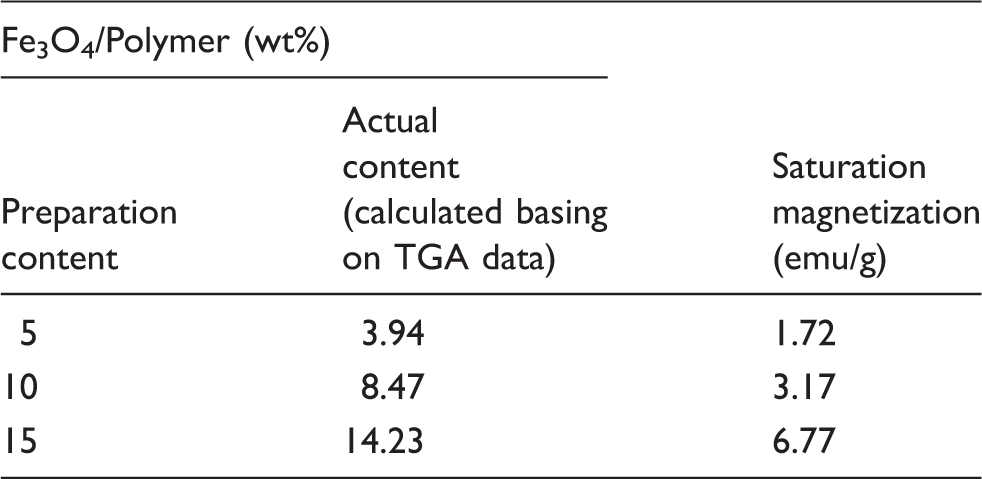
As a magnetic material, the magnetic property of the Fe3O4/polymer nanofiber nonwoven is important to its applications. The magnetization curves at 25°C of the Fe3O4/polymer nanofiber nonwoven are shown in Figure 7a. The results in Table 2 show that the magnetic nanofiber nonwoven has saturation magnetization of 1.72 emu/g to 6.77 emu/g, which is lower than that of the corresponding bulk (92 emu/g)
46
resulting from the existence of polymer.
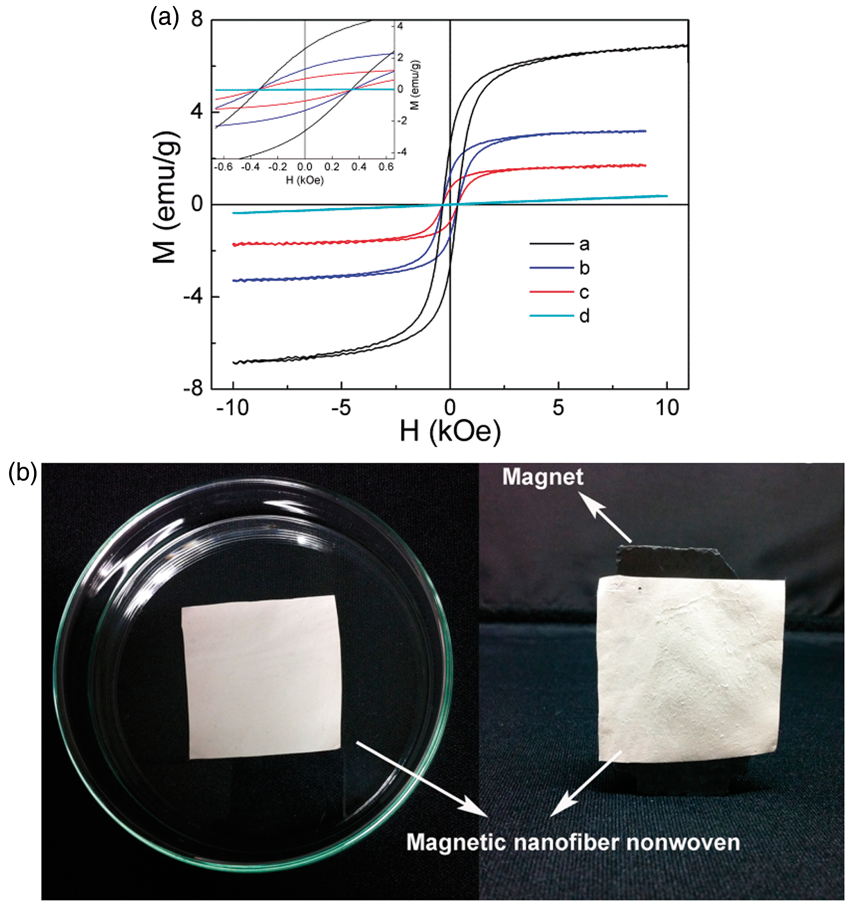
The coercivity is also about 300 Oe at 25°C (see the inset of Figure 7a). Therefore, the products show a soft ferromagnetic behavior, which is the same as Fe3O4. The magnetism of the products was demonstrated by holding a commercial magnet close to the sample as shown in Figure 7b. The magnetic nanofiber nonwoven was attracted to the magnetite, indicating that the nanofiber nonwoven had strong magnetism.
Water resistance of Fe3O4 NPs/PAA/PVA composite nanofibers
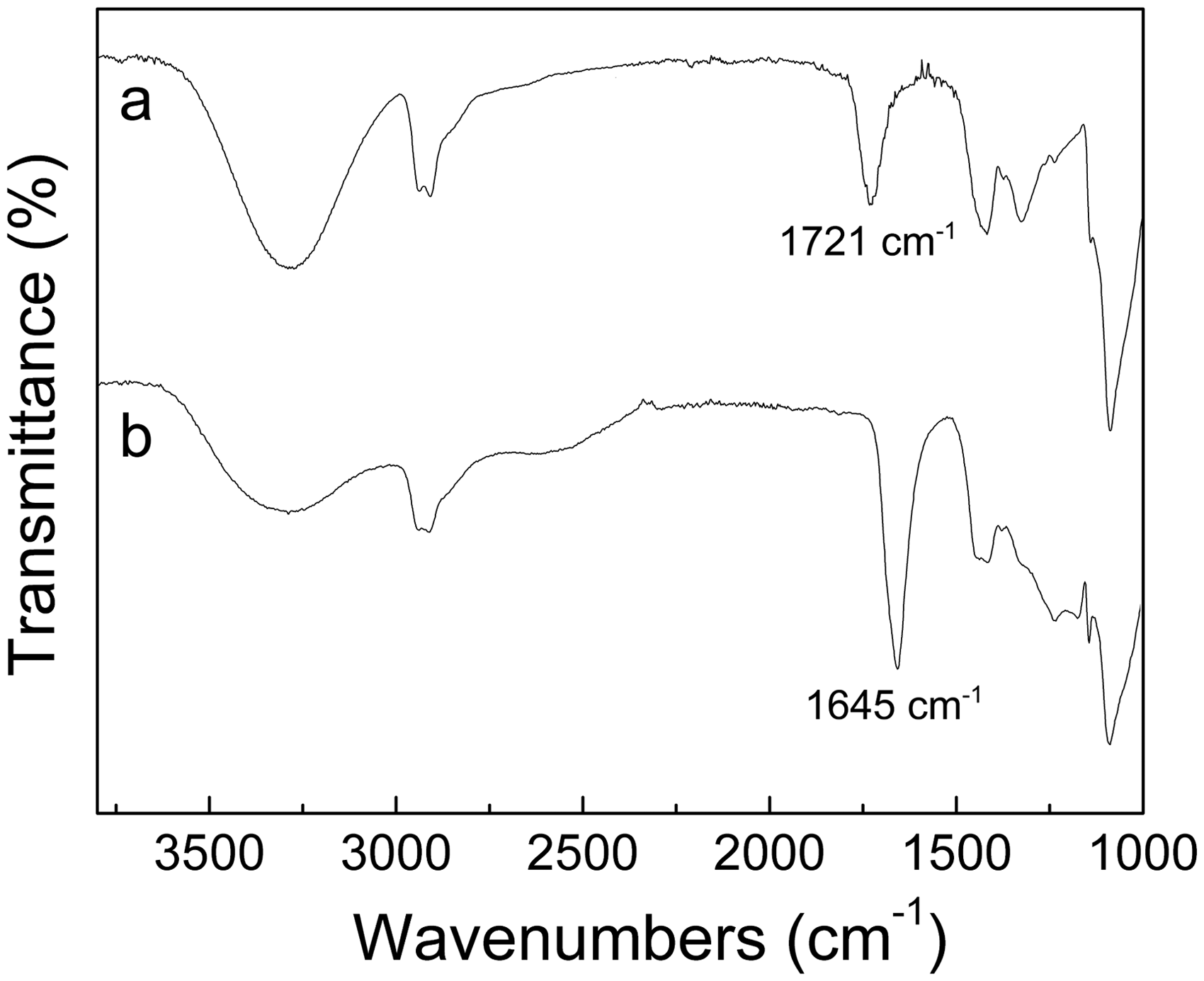
The biggest drawback of electrospun PVA nanofibers, and a significant limit to their practical application, is the poor stability in water of the material.47,48 To improve the water resistance of PVA nanofibers, most researchers have concentrated on chemical cross-linking by a variety of substances including glutaraldehyde, acetaldehyde, or formaldehyde.49,50 In our study, PAA was used not only as a dispersing agent for Fe3O4 NPs, but also as a cross-linking agent to enhance the water resistance of PVA composite nanofibers. Figure 8 shows SEM images of Fe3O4 NPs/polymer composite nanofibers after being immersed in water for several days. The Fe3O4 NPs/PVA nanofibers quickly swelled and the unique nanofibrous structure is lost by dissolution into water. However, the Fe3O4 NPs/PAA/PVA nanofibers only swelled and maintained their fibrous structures after being in contact with water for 1 d. This indicates that chemical cross-linking between PVA and PAA that maybe occurred through an ester formation between the –OH groups in PVA and the –COOH groups in PAA during heat treatment at 100°C for 2 h, which was confirmed by the Fourier transform infrared (FT-IR) analysis (Figure 9). Compared to the FT-IR spectrum of composite fibers before heat treatment, the ones of heat-treated PVA/PAA composite fibers (PAA/PVA = 1:1) shows a less amount of –OH groups (3400 cm-1) left in the polymers. Furthermore, it is clearly seen that the –C=O stretching vibration at 1645 cm−1 becomes stronger after heat treatment. This indicates that the –C=O stretching vibration (1721 cm-1) is normally shifted to lower frequencies through an ester (–(C=O)O) formation between the –OH groups in PVA and the –COOH groups in PAA.
39
SEM images of Fe3O4/polymer nanofibers after contact with water for different periods, followed by vacuum drying. Fourier transform infrared spectra of Fe3O4 NPs/PVA/PAA composite fibers (PAA/PVA = 1:1), 
Conclusion
Fe3O4 NPs/PAA/PVA composite nanofibers were prepared via the electrospinning of PAA/PVA aqueous solutions with magnetite Fe3O4 NPs which were homogenously dispersed by PAA. Experiment results showed that the diameters of Fe3O4 NPs were in a range of 10 nm to 40 nm under our experimental conditions; Fe3O4 NPs stabilized by PAA can be homogenously dispersed in PVA composite nanofibers; the crystallinity of PAA/PVA nanofibers increased along with the increase in Fe3O4 NPs content. The magnetic property of Fe3O4 NPs/PAA/PVA composite nanofibers was also investigated and the results showed that the composite nanofibers show a strong magnetism and soft ferromagnetic behavior which is the same as Fe3O4 at room temperature. In addition, the water resistance of Fe3O4 NPs/PAA/PVA composite nanofibers was enhanced by heat treatment. As a result, in our process to preparing magnetic Fe3O4 NPs/PVA composite nanofibers, PAA acts not only as a dispersant but also as a cross-linking agent. This method provides a simple and convenient process to prepare homogenous NPs/PVA composite system with high stability and can greatly improve the practical application of NPs/PVA composite.
Footnotes
Funding
This work was supported by Grant-in-Aid for Global Center of Excellence (COE) Program by the Ministry of Education, Culture, Sports, Science, and Technology, Japan.