
Abstract
Haemophagocytic lymphohistiocytosis (HLH) is a rare and life-threatening hyper-inflammatory disorder, resulting from an uncontrolled activation of macrophages and cytotoxic T cells and leading to a cytokine storm accompanied by multi-organ dysfunction.
Introduction
HLH is caused by defective regulation of immune responses, resulting in excessive activation of macrophages and cytotoxic T cells. 1 It may be primary (familial), typically due to genetic mutations affecting cytotoxic granule-mediated lymphocyte function or secondary, triggered by infections, malignancy or autoimmune disorders. The incidence of familial HLH (FHL) is estimated as 1 in 50,000–100,000 live births, with early infancy representing the most vulnerable period. 2 Clinically, HLH manifests with prolonged fever, hepatosplenomegaly, cytopenia, coagulopathy and multi-organ dysfunction, often mimicking severe sepsis or metabolic disorders. Early diagnosis is challenging, particularly in neonates and young infants, owing to non-specific features and limited access to genetic testing in many centres, especially in low-income countries. Histopathology and autopsy remain invaluable tools for confirming diagnosis, documenting the extent of organ involvement and enhancing understanding of disease pathogenesis.
Case report
An 11-week baby boy, born at 38 weeks’ gestation by normal vaginal delivery with a birth weight of 2.47 kg, presented with intermittent fever, progressive abdominal distension, pallor, icterus, pedal oedema and extensive vesicular and macular rash over three weeks. His perinatal, developmental and vaccination history was unremarkable, and there was no family history of haematological nor auto-immune disorder, nor consanguinity. However, his mother had a turbulent obstetrical history, including one spontaneous abortion in the first trimester and one stillbirth.
On admission, the infant was febrile (38.5°C), tachycardic (pulse 128/min), hypotensive (BP 84/48 mmHg) but not hypoxic (oxygen saturation of 98% on room air). Examination revealed generalised pallor, icterus and petechiae at prior cannulation sites and hepatosplenomegaly (liver palpable 7 cm, spleen 2.5 cm below respective costal margins) (Fig. 1(a) and (b)).
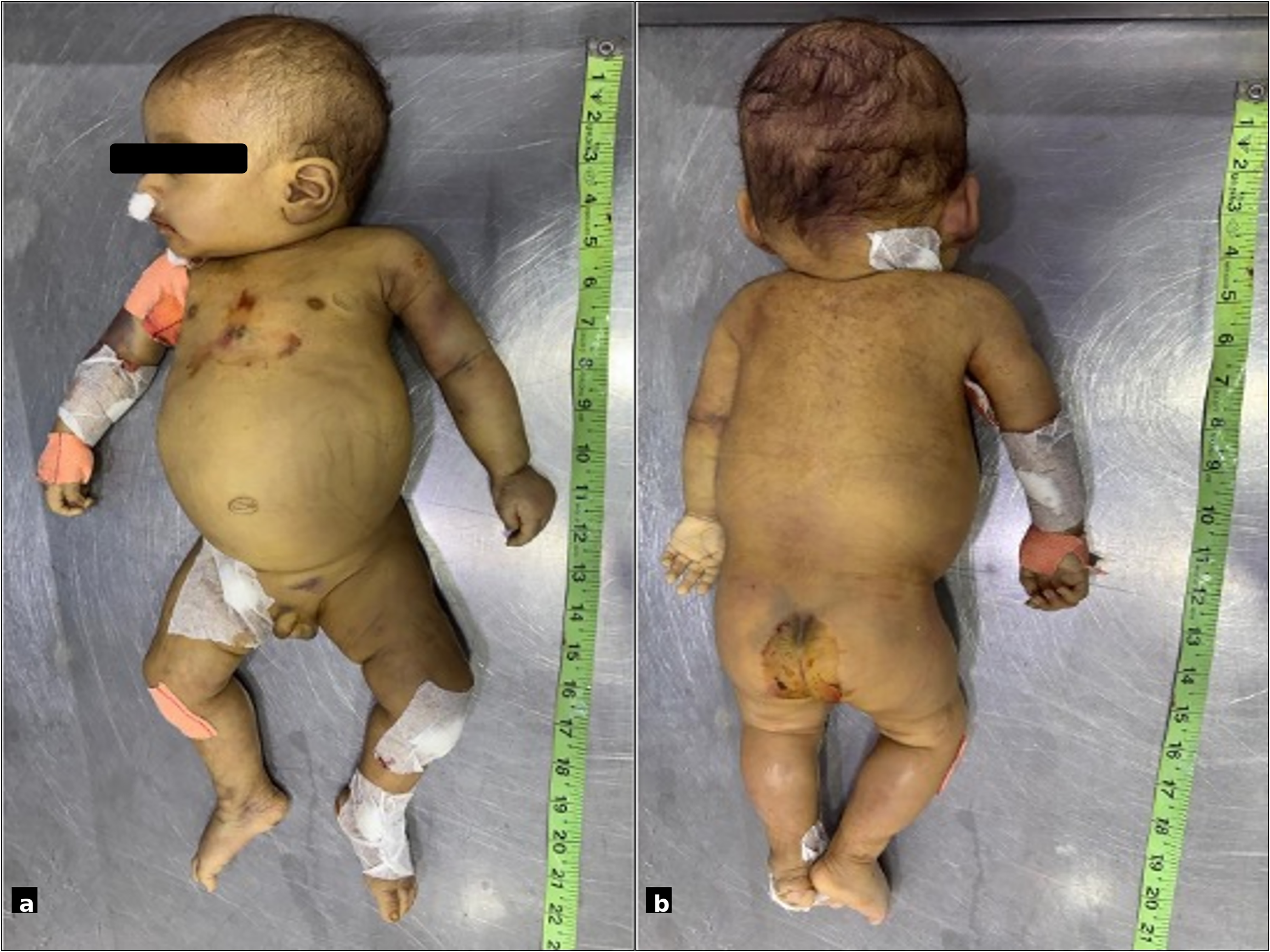
(a and b) Clinical images demonstrating hepatosplenomegaly with generalised pallor, icterus and petechial haemorrhages at previous cannulation sites.
Laboratory investigations showed anaemia (Hb 102 g/L), leucocytosis (11.7 × 109/L, neutrophils 66%, lymphocytes 25%) and severe thrombocytopenia (platelets 25 × 109/L). Coagulation studies were markedly deranged (PT/aPTT >320/>500 s) with elevated
A provisional diagnosis of HLH with hepatic dysfunction and disseminated intravascular coagulation was made. Management included intravenous antibiotics (piperacillin–tazobactam and amikacin), dexamethasone, vitamin K and transfusions of platelets and fresh frozen plasma. Despite these efforts, severe hypoglycaemia (<1.4 mmol/L) ensued and subsequently apnoea, bradycardia and cardiovascular collapse. Resuscitation efforts were unsuccessful, and the infant demised. The interval between hospital admission and death was c. 22 h.
Autopsy demonstrated a weight of 4.5 kg, crown–heel length of 52 cm, with normally sited thoraco-abdominal organs. The thymus (4 × 3 × 1 cm, 3.2 g) showed involution with crowded Hassall's corpuscles. The heart (105 g) revealed a small left atrial clot; the lungs were congested and consolidated with subpleural haemorrhage and numerous haemophagocytic histiocytes. The liver (14 × 10 × 4.5 cm, 279 g) exhibited necrosis, sinusoidal dilation and haemophagocytosis. The spleen (7 × 5 × 3 cm, 30 g with pancreas) had sinusoidal histiocytic proliferation. Multiple lymph nodes, adrenals and stomach showed infiltration by haemophagocytic cells. The kidneys showed acute tubular necrosis; but pancreas and intestines were unremarkable. Representative microscopic findings from the involved organs are depicted in Figures 2(a) to (d) and 3(a) to (d). Molecular analysis confirmed FHL2.

Histopathological features of haemophagocytic lymphohistiocytosis (HLH) involving multiple organs. (a and b) Liver demonstrating disrupted hepatic architecture with focal submassive hepatocyte necrosis, bridging necrosis and clusters of large histiocytes exhibiting haemophagocytosis (H&E, 100×, 200×); (c) Spleen showing effacement of normal splenic architecture by diffuse sheets of large histiocytes with prominent haemophagocytosis, involving both red and white pulp (H&E, 100×); (d) Adrenal glands showing focal sheets of HLH cells causing partial parenchymal effacement, with adjacent preserved adrenal tissue (H&E, 200×).

Histopathological features of haemophagocytic lymphohistiocytosis (HLH) involving multiple organs. (a and b) Stomach showing near-complete effacement of mucosa and submucosa by dense infiltrates of histiocytes with haemophagocytosis, with loss of normal glandular architecture (H&E, 40×, 400×); (c) Pancreas with preserved acinar lobular architecture; adjacent lymph nodes completely replaced by sheets of haemophagocytic histiocytes (H&E, 40×); (d) Lung showing haemorrhage with focal hyaline membrane formation with presence of abundant haemophagocytic histiocytes (H&E, 200×).
Discussion
This case demonstrates the fulminant course and extensive multisystem involvement of HLH in early infancy. 3 Thymic involution indicated chronic immune stress, while widespread hepatic necrosis and sinusoidal histiocytosis highlighted severe cytokine-driven hepatocellular injury. Pulmonary consolidation and haemorrhage probably contributed to terminal cardiopulmonary failure.
Diagnosis relies on the HLH-2004 criteria in children, with the HScore serving as a complementary tool for estimating probability in atypical or adult cases.4–7 Haemophagocytic lymphohistiocytosis is broadly classified as primary or secondary based on the presence of underlying genetic defects. Primary HLH arises from mutations affecting cytotoxic T-cell and natural killer cell function, including PRF1, SH2D1A and LYST. 8 Early genetic testing is critical, as allogeneic hematopoietic stem cell transplantation remains the definitive therapy for pHLH. Secondary HLH occurs as a reactive syndrome triggered by infection, malignancy or auto-immune or auto-inflammatory conditions, described as a macrophage activation syndrome, frequently observed in systemic juvenile idiopathic arthritis, systemic lupus erythematosus, Kawasaki disease and adult-onset Still's disease. Establishing a diagnosis then requires exclusion of pathogenic mutations in HLH-associated genes.
Compound heterozygous mutations, leading to persistent immune activation and rapid progression to multiorgan failure, demand early recognition and prompt treatment with dexamethasone, etoposide and cyclosporine. In the case described, informed family counselling regarding recurrence risk could then be provided.
Autopsy remains indispensable for definitive diagnosis, detailed documentation of multiorgan involvement and expanding understanding of disease mechanisms in rapidly fatal presentations.
Footnotes
Ethical statement
For this report, formal consent from a local ethics committee was not required.
Patient consent
The authors certify that they obtained appropriate consent from the patient's legal guardians. The guardians understand that names and initials will not be published and that due efforts have been made to conceal identity.
Author contributions
Asit Ranjan Mridha and Md Ali Osama conceptualised the study. Sanjana Goyal and Nikita Jakhar curated the data. Project administration was carried out by Asit Ranjan Mridha. The original draft was prepared by Md Ali Osama. All authors contributed to reviewing and editing and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
All data generated or analyzed during this study are included in this published article. Further enquiries can be directed to the corresponding author.
Guarantor
The corresponding author is the guarantor of submission.
Statement of human and animal rights
This study involved human participants and was conducted in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments. No animal studies were conducted.