
Abstract
In this chapter, an overview of some of the prominent risk factors that contribute to the pathophysiology of venous thrombosis will be discussed. In 1856, Dr Rudolf Virchow developed the concept outlining the genesis of intravascular thrombosis. Dr Virchow hypothesized that circulatory stasis due to interrupted blood flow, changes in the blood leading to blood coagulation, and irritation or damage to the vascular endothelium would initiate acute venous thrombus generation. Presently, it is known that these above-mentioned risk factors are influenced by increasing age, gender, and obesity. The current chapter will focus on recent preclinical and clinical investigations that will give the reader insight into the prothrombotic mechanisms that lead to acute venous thrombosis.
Introduction
Venous thromboembolism (VTE), which includes deep venous thrombosis (DVT) and pulmonary embolism (PE), is a significant health problem that leads to severe morbidity and mortality. Despite improvements in prophylaxis and treatment of VTE, the epidemiology of this disease has not changed significantly over the past 30 years.
Risk factors for acute venous thrombosis (VT)
The factors that could lead to or interact with one another to lead to VTE were first described by Rudolf Virchow in 1856 and include (1) blood stasis, (2) endothelial injury, and (3) blood hypercoagulability.
1
Of these three factors, vein wall endothelial damage triggers a local inflammatory response, which promotes a prothrombotic state driven by tissue factor (TF), adhesion molecules, hemostatic factors, and proinflammatory cytokines which support acute VTE. In 1974, it was first hypothesized that vascular inflammation and thrombosis are interrelated. The original hypothesis suggested that prothrombotic factors lead to the activation of leukocytes and platelets. This process promotes thrombus amplification via adherence and layering of the activated platelets and leukocytes. This hypothesis is supported in current vascular research findings. However, a number of risk factors that can lead to the development of VTE have been identified in both animal and human studies which include aging, gender, and obesity will be covered in detail in this chapter (Figure 1).
Risk factors for acute venous thrombosis as it relates to Virchow’s triad.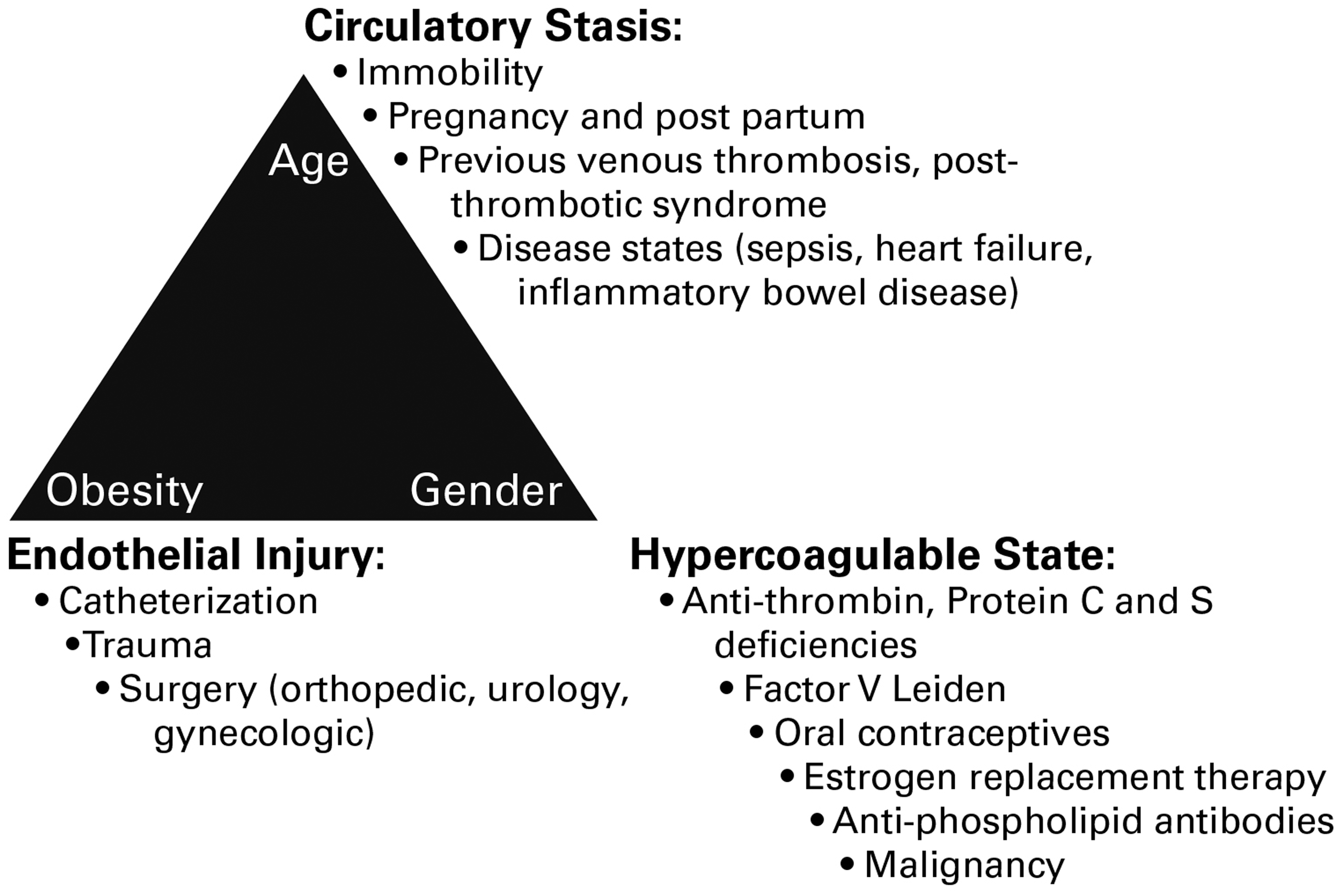
Aging and VT
The aging process is an important common denominator in the development of VTE. The incidence of VT is 2–7 times higher in those greater than 55 years of age as compared to younger aged groups, and the incidence increases 74% per decade of age over 45 years. In a community-based study of phlebographically documented VT, the yearly incidence of VT was noted to increase progressively from almost 0 in childhood to 7.65 cases per 1000 in men and 8.22 cases per 1000 in women older than 80 years. 2 The incidence of VT increased 30-fold from those aged 30 years to those older than 80 years. Furthermore, Hansson et al. found the prevalence of objectively documented thromboembolic events among men to increase from 0.5% at age 50 to 3.8% at age 80 years. 3 The influence of age on the incidence of VTE is likely to be multifactorial. The number of thrombotic risk factors increases with age, three or more risk factors being present in only 3% of hospitalized patients younger than 40 years but present in 30% of those 40 years and older. 4 Interestingly, it also appears that the number of risk factors required to initiate thrombosis decreases with age. This may be related to an acquired prothrombotic state associated with aging as higher levels of thrombin activation markers are found among older people. Advanced age also has been associated with anatomic changes in the soleal veins and more pronounced stasis in the venous valve pockets. 5 Thickening of venous valve cusps due to increases in valve collagen and intimal thickening with age has also been documented.
Previous work from our laboratory and others, support the working hypothesis that with aging, P-selectin surface expression from vein wall (Weibel--Palade bodies, WPBs) and platelets (a-granules) increases, thus producing a procoagulant environment with activated platelets in blood quickly associating with leukocytes. These activated leukocytes roll and tether on the stimulated vein wall, promoting greater thrombus formation than noted in younger individuals.
6
(Figure 2).
Aging increases activation of P-selectin and proinflammatory cytokine release for the venous endothelium. This initiates platelet activation which elicits a cycle of injury from vein-to-platelet-to-vein that is influenced significantly by age. Activated platelets release P-selectin that promote leukocyte attachment and incorporation into a developing thrombus at the point of vascular injury. The physical presence of the thrombosis initiates inflammatory signaling that increases the release of more P-selectin and inflammatory mediators to continue this vicious cycle. IL-1: Interleukin-1; IL-6: Interleukin-6; PSGL-1: P-selectin glycoprotein ligand-1; P-Sel: P-selectin; SP-sel: Soluble P-Selectin; TF: Tissue Factor; TNF: Tumor Necrosis Factor; VWF: Von Willebrand Factor.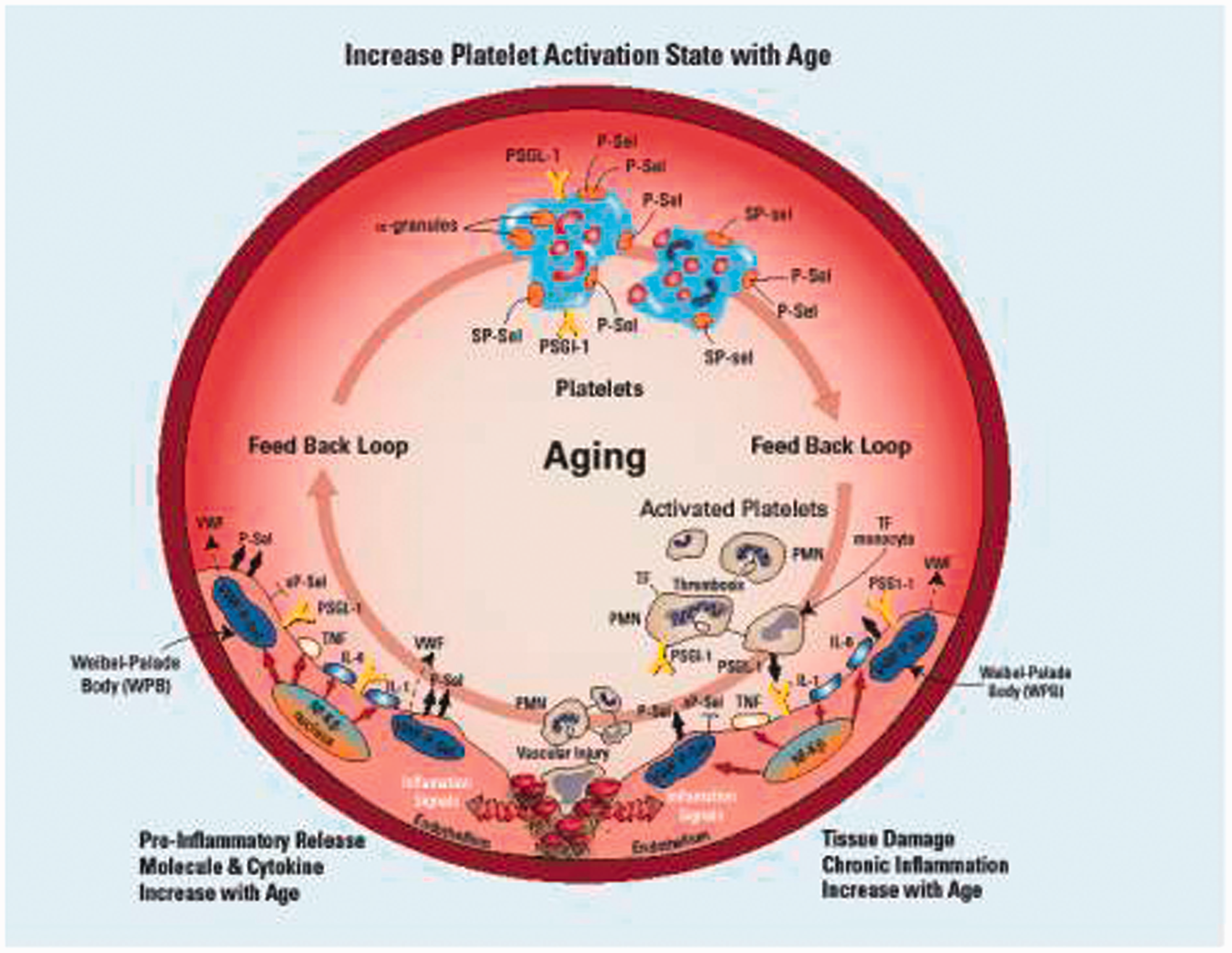
Gender and VT
The role of gender in scientific studies is becoming a significant issue. 7 In fact, NIH is considering policies “to include, compare, and contrast experimental findings in male and female animals and cells.” In this light, we have characterized differences in VT in our mouse model related to gender and propose to study gender as it relates to VT. Male mice had significantly larger venous thrombi than females. Using the nonocclusive thrombus EIM model, we studied male and female C57BL/6 mice (6–8 weeks) who underwent inferior vena cava thrombosis. In aged-matched groups, we found that male VT were significantly larger than female VT at days 2 (13.1 ± 1.0 versus 6.8 ± 0.5 × 10−3 g, p < 0.01), 6 (10.4 ± 0.8 versus 5.4 ± 0.5 × 10−3 g, p < 0.01), and 14 (6.3 ± 0.5 versus 4.1 ± 0.3 × 10−3 g, p < 0.01). 8 In the literature, there is controversy with some studies suggesting a slightly higher risk of VT in men than women, while others suggest the opposite, consistently males have been shown to have a higher rate of recurrent VT. 9 Results from a large population based case–control study suggest men not only have a higher risk of recurrent VT, but also a 2.1-fold increased risk of first VT, when women are analyzed without reproductive risk factors being involved.
Obesity and VT
Obesity is an epidemic in the United States. A recent survey in the United States reported that 30.5% of adults are obese (body mass index (BMI) > 30 kg/m2) and 64.5% are considered overweight (BMI >25 kg/m2).10,11 This trend is even evident in adolescents as 15.5% of children between 12 and 19 years of age are now considered overweight. Current estimates by the World Health Organization predict that there will be 700 million obese individuals worldwide by the year 2015. Importantly, it is also apparent from clinical studies that excess visceral adiposity with metabolic syndrome is a risk factor for VT, suggesting a direct effect of central obesity on thrombus initiation. 12 In a case–control study of 150 normal individuals and 146 with an objectively defined first-time VT, abdominal obesity was found to be associated with the highest VTE risk, and further adjustment for the presence of thrombophilic states did not change this result. Further clinical studies demonstrated that elevated bodyweight is associated with a higher prevalence of idiopathic VT and PE and that it is also correlated with an increase in the recurrence rate of VTE. 13 In the mouse model of diet-induced obesity, increased body weight has been found to be associated with VT.
Studies with confocal laser microscopy have revealed increased interactions between leukocytes, endothelial cells, and platelets in the microcirculation of visceral adipose tissue in genetically and diet-induced obese mice and demonstrated signs of local platelet activation, such as increased P-selectin expression and the formation of monocyte–platelet aggregates. It has also been shown that genetic deletion of the receptor for P-selectin and E-selectin (P-selectin glycoprotein ligand 1 (PSGL-1)) in mice abolishes obesity-induced increased leukocyte–endothelial interactions and reduces the macrophage infiltration into visceral fat depots. 14
Obesity is associated with increases in fibrinogen, factor VII, plasminogen activator inhibitor (PAI-1), and changes in platelet function. Additionally, specific adipokines mediate thrombosis, such as leptin (promoting platelet adhesion, activation, and aggregation and increasing TF expression), C-reactive protein (promoting P-selectin expression), and a reduction of adiponectin in obesity (a natural antithrombotic). 15 PAI-1, released from adipose tissue and the liver, is up-regulated in obesity and mice expressing elevated PAI-1 have been shown to produce enhanced VT. In mice aged 8–10 weeks, we have demonstrated in preliminary data that those with genetically induced hyperlipidemia via deletion of Apolipoprotein E (ApoE–/–) weighed more on average (26 g versus 24 g, p < 0.001), compared to WT mice. We have previously shown that ApoE–/– animals form larger venous thrombi and exhibited impairment of the uPA-tPA-plasmin axis via up-regulation of PAI-1. 16
The pathophysiology of VT
The vascular inflammatory response is initially protective by nature; its role is to promote the recruitment of inflammatory cells for the removal of micro-organisms and endotoxins. However, local and systemic inflammation can produce a prothrombotic environment driven by platelet activation, TF, prothrombotic microparticles (MPs), neutrophil extracellular traps (NETs), adhesion molecules, hemostatic factors, and proinflammatory cytokines. Various research laboratories have used multiple animal models of VT to dissect out the mechanisms regulating thrombus formation and thrombus resolution.
Inflammation and thrombosis have been shown to interact and share common mechanisms. Vascular injury and various disease states can promote the exposure of TF within the vessel wall to blood flow, thus leading to the initiation of thrombosis. TF is a membrane-bound protein (46 kDa) that triggers thrombin generation by forming a complex with factor VIIa, triggering the activation of the coagulation cascade. TF pathway inhibitor is the endogenous polypeptide that acts to reversibly inhibit formation of both thrombin and factor Xa. Currently, the exact distribution of TF and which source (vein wall versus cellular components) is most significant in promoting a prothrombotic environment is under investigation. In fact, both sources may be important and the overall effect may depend on the nature of the stimulus. Vein wall TF may be most important with vein wall injury or vein wall dilatation, while leukocyte-derived TF may be most important when there is no vein wall injury. Work in our laboratory has demonstrated that with vein wall injury and exposure of vein wall TF, this TF is more important in the thrombogenic process than the TF that is brought to the point of thrombogenesis by activated MPs.
P- and E-selectin are cell adhesion molecules with critical roles in thrombogenesis, as shown in numerous animal studies. Investigations using rat and mouse thrombosis models demonstrated up-regulation of P-selectin and E-selectin in the vein wall 6 h and six days after thrombus induction, respectively. The increase in the number of P-selectin molecules present on the endothelial cell surface is due to their release from the WPB. WPBs are the endothelial-specific storage organelles for regulated secretion of Von Willebrand Factor (vWF) and P-selectin onto the endothelial cell membrane. Thus, the exocytosis of WPB initiates a rapid translocation of P-selectin to the activated endothelial surface resulting in P-selectin-receptor binding (PSGL-1) to both leukocytes and platelets.
The therapeutic benefits of P-selectin inhibition of acute VT has been well documented. Using both rodent and nonhuman primate animal models, the relationship between selectins and thrombosis has been defined. In animal models, P-selectin inhibition, with either an antibody, small molecule inhibitor, or aptamer administered prophylactically or post-VT, significantly decreased thrombogenesis, decreased vein wall fibrosis, and increased venous thrombus resolution in these animal models. Previous studies have demonstrated the beneficial effects of combined P- and E-selectin inhibition in decreasing DVT in a mouse model of stasis thrombosis. E-selectin is expressed on activated vascular endothelium; there are several receptors for E-selectin with E-selectin ligand-1 and CD44 being primary. Published work shows that mice gene deleted for both E- and P-selectin had significantly smaller venous thrombi versus control animals in a stasis model of VT. Mice gene deleted for E-selectin had the lowest thrombus burden, evaluated two and six days post thrombosis, suggesting that E-selectin inhibition could be an effective therapeutic strategy for VT.
MPs are involved in the thrombotic process and the amplification of thrombosis. MPs are small (less than 1 µm, standardization of MP measurement is still the topic of ongoing research), phospholipid vesicles, which are shed from platelets, leukocytes, and endothelial cells in a calcium-dependent fashion. MPs are a normal constituent of blood and can be isolated from plasma by ultracentrifugation. MPs lack DNA but recent evidence suggests they may carry RNA. The P-selectin receptor, called PSGL-1, is expressed on leukocytes and platelets, as well as on their derived MPs. Upon vascular injury, E-selectin is temporally expressed on activated vascular endothelium after P-selectin. MPs coexpressing TF and leukocyte markers have been shown to accumulate in growing thrombi in a P-selectin: PSGL-1-dependent fashion. P-selectin: PSGL-1 interactions also stimulate the production of thrombogenic MPs from leukocytes, particularly monocytes along with platelets and endothelial cells. These prothrombotic MPs express TF and possess a phosphatidylserine-rich anionic surface capable of assembling complexes of the coagulation cascade. MPs and inflammatory cells are recruited back into the area of thrombus formation leading to thrombus amplification (Figure 3).
Proposed mechanisms of acute venous thrombosis postactivation of the venous endothelium. The activation of tissue factor, P-selectin and E-selectin, platelets, inflammatory cells, microparticles, and the formation of neutrophil endothelial traps (NETs), lead to acute thrombus formation and amplification. CD44: Cluster of differentiation 44; ESL-1: E-selectin ligand; MP: microparticles; NETs: neutrophil endothelial traps; PSGL-1: P-selectin glycoprotein ligand-1; VWF: Von Willebrand Factor.
MPs also express on their surface PSGL-1, which then can bind to up-regulated P-selectin on platelet surfaces in the thrombus, allowing for the MPs to be concentrated in the area of the developing thrombus MPs reinjected into hemophilic mice, normalized their tail bleeding times, and human pericardial-derived MPs expressing TF have been demonstrated to increase thrombosis in a rat venous stasis model. The importance of P-selectin: PSGL-1 to VT likely depends on the nature of the stimulus and the role of TF, which is normally abundant in the outer portion of the vein wall. PAI-1 is stored in the α-granules of quiescent platelets. PAI-1 is a potent inhibitor of tissue plasminogen activator and urokinase which are largely responsible for the initiation of fibrinolysis. On activation, MPs shed from platelets express PAI-1 and are localized to the growing thrombus via P-selectin: PSGL-1 interactions. Therefore, platelet-derived MPs are not only prothrombotic but also inhibit fibrinolysis, delaying thrombus resolution and facilitating thrombus growth.
The role of NETs in acute inflammation has been demonstrated. Activated neutrophils have been shown to release chromatin DNA and histone-containing granular antimicrobial proteins which form extracellular matrices or traps, this in addition to their phagocytic and bactericidal functions. Specifically, it has recently been shown that activation of venous endothelium generates signals that lead to leukocyte movement, adhesion, and activation. Through processes that also involve the initial activation of platelets and leukocytes, neutrophils polymorphonuclear cell (PMN) initiate and amplify thrombosis through the formation of NETs, which are extracellular fragments of DNA containing histones and antimicrobial proteins (Figure 3). In vitro and in vivo, NETs provide a scaffold and stimulus for thrombus formation. 17 NETs bind adhesion molecules such as VWF and fibrinogen, promote fibrin formation, facilitate platelet aggregation, and stimulate recruitment of red blood cells. Plasma extracellular DNA is present at the time of thrombus formation in murine, baboon and human DVT samples.17,18,19 PMNs may also serve to amplify thrombus via heterotopic interactions with red blood cells and platelets.
Inflammatory cells are important to the process of thrombus recanalization and organization. In a study using an antibody to neutrophils, animals rendered neutropenic developed significantly larger venous thrombi. Of interest, neutropenic cancer patients are not protected from VT, and multiple neutropenic episodes are significantly associated with recurrent VTE in patients with malignant disease who require filter placement due to a failure of, or contraindication to, anticoagulation. In a previous study using a well-characterized mouse inferior vena cava (IVC) ligation model of VT, it was demonstrated that neutrophil vein wall inflammatory cell influx occurs acutely and was then followed by a later influx of monocytes during thrombosis. This inflammatory cell extravasation process has also been documented in multiple animal models of VT. Neutrophils are necessary for early thrombus resolution by promoting the lysis of both fibrin and collagen (Figure 4). Inflammatory cells, especially monocytes, have been shown to promote thrombolysis due to their secretion of proteolytic enzymes. Postthrombosis, increasing levels of monocyte chemotactic protein-1 (MCP-1) stimulate chemotaxis and activation of monocytes which promote venous thrombus resolution.
Inflammatory cells in a two-day mouse venous thrombi. Two days postvenous thrombosis in a mouse, note the normal events of inflammatory cells, predominately neutrophils, recruited to the vein wall (V)–thrombus (T) interface. Paraffin embedded, stained with hematoxylin and eosin (H&E), magnification (40X).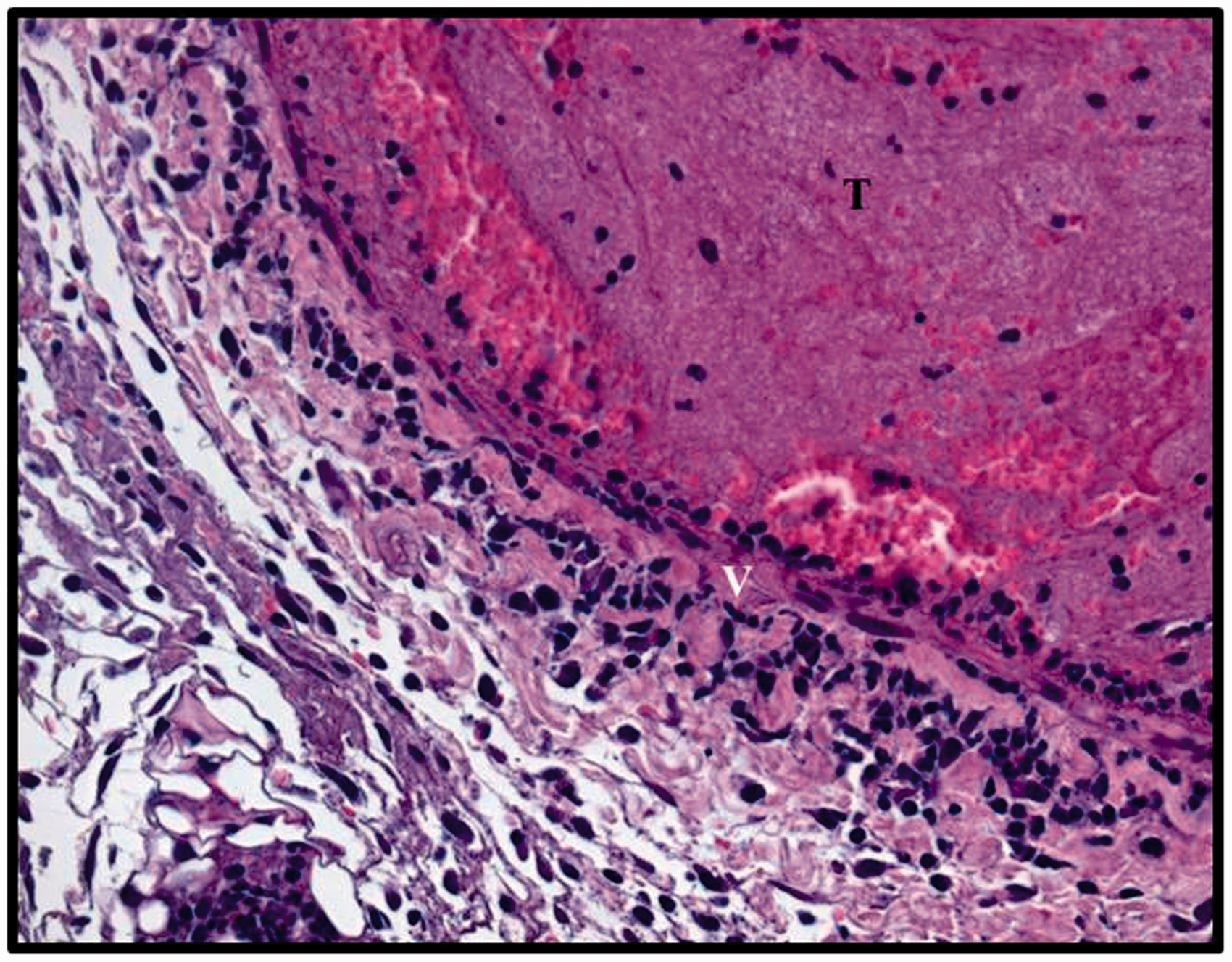
Summary
Venous thrombi are associated with significant acute inflammatory responses which can promote thrombus propagation by creating a prothrombotic environment. The development of venous thrombi can be strongly influenced by aging, gender, and obesity. It is imperative that preclinical and clinical research efforts continue to define the disease mechanisms that regulate DVT in order to develop personalized treatment strategies that improve patient outcomes.
Footnotes
Acknowledgements
The author would like to thank Dr Thomas W. Wakefield for his friendship, support, mentorship, and tireless commitment to the treatment of VT.
Conflict of interest
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.