
Abstract
Vascular malformations are abnormal growth or development of the vascular structure that result from genetic mutations during early vascular development. Traditional invasive treatment for vascular malformations includes embolo-sclerotherapy, cryotherapy, laser therapy and surgery. However, surgical or minimally invasive treatment is rarely optimal due to the risk of treatment complications, and a complete cure is often difficult to achieve. Targeted therapy can be guided by the current understanding of molecular signalling pathways and disease classifications. Existing and novel medical treatments target the major cellular signalling pathways implicated in the pathogenesis of vascular malformations: mTOR inhibitors, phosphatidylinositol-4,5-biphsophate 3-kinase catalytic subunit alpha (PIK3CA) inhibitors, and AKT inhibitors are being developed to target the Phosphoinositide 3-Kinase (PI3K)/AKT/mTOR pathway, while mitogen-activated protein kinase (MEK) inhibitors and BRAF inhibitor are being researched to target the RAS/RAF/MEK/ERK pathway. Angiogenesis inhibitors are also utilised in the treatment of vascular abnormalities. This review aims to discuss the evolving medical therapy available in the treatment of Vascular Malformations.
Keywords
Introduction
Vascular malformation is a subgroup of vascular anomaly characterised by abnormal growth or development of blood or lymphatic vessels. 1 Lesions are derived from aberrant embryonic development of vascular channels and feature dysplastic vessels. In the morphological development of the vascular system, genetic mutations cause developmental errors that result in vascular malformations. 2 Vascular malformations consist primarily of abnormal blood or lymphatic vessels lined by normal endothelial cells, which do not undergo abnormal cellular turnover. They are typically present at birth, grow proportionally with the patient, and do not spontaneously regress or disappear. They may also develop disproportionately during growth spurts and puberty as a result of hormonally induced dilation or expansion of already abnormal vessels.2–4 Depending on the anatomical location and affected structures, lesions can manifest as focal, multifocal, or diffuse with varying appearances, symptoms, and severity. Pain, deformity, and functional impairment are the prevalent symptoms.2,3 In addition, they can cause local complications, such as bleeding and infection, as well as systemic complications, such as ischemia, thromboembolism, and organ failure. Due to its chronic pathological characteristics, vascular malformations can cause emotional distress, and a reduced quality of life compared to the general population.5,6
The International Society for the Study of Vascular Anomalies (ISSVA) classifies vascular malformation into five groups: simple, combined, anomalies of major named vessels, malformations associated with other anomalies, and provisionally unclassified vascular anomalies.3,7–9 In addition, vascular malformations can be described based on their flow characteristics and further subclassified into four major categories according to the type of blood vessels. Low-flow vascular malformations include Venous Malformation (VM), capillary malformation (CM) and lymphatic malformation (LM), and a combination of them. High-flow vascular malformations include arteriovenous malformation (AVM). 1
Vascular malformations continue to present a therapeutic challenge due to their unpredictability, treatment response, and inevitable recurrence. In the past, therapeutic options for the treatment of vascular malformation were limited, and surgical management was regarded as the initial treatment option. However, the lack of understanding of the pathogenesis of vascular malformations has led to poor outcomes.
Understanding the genetic mutations improves our knowledge of the relevant signalling pathways lead to the pathogenesis of vascular malformations. Signalling pathways are series of reactions, involving a set of genes which code for specific proteins to carry out a specific function. Gene mutations gene on this pathway can lead to a change in part of the molecular functioning of this pathway. Therefore, understanding the mutations which cause vascular malformations contributes to the understanding of the underlying molecular mechanisms. 10 Pharmacological therapies can target these specific proteins in the pathway to treat vascular malformations. Therefore, greater understanding of these mechanisms can lead to greater opportunities in identifying potential new targeted pharmacological therapies. 11
Current treatment options and potential new therapeutic agents for vascular malformations are discussed in this article.
This study aims to review the current medical treatment for vascular malformations and treatments that are being investigated.
Methods
A literature search was performed on Embase (1980-2022) and Medline (1980-2022). The MeSH terms used in Embase were “vascular malformation”, “medication”, and “drug”. The MeSH terms used in Medline were “vascular malformation”, “medication”, and “drug”. Only original research articles in English which reported on vascular malformations were included, and unpublished material, abstracts and letters were all excluded.
Results
Molecular signalling pathway in vascular malformations
Vascular malformation is a mosaic disorder resulting from inherited or somatic genetic mutations that cause abnormal signalling within vascular endothelial cells.3,12–14 Mosaic disorders are where somatic, de novo mutations result in cells having variations.
15
The majority of vascular malformations are caused by monogenetic mutation, either as a result of sporadic somatic mutation alone or the loss of a second allele in a pre-existing germline mutation, which can lead to the progressive development of multifocal lesions.
14
Typically, vascular malformation mutations occur in genes encoding proteins involved in one or more of the two major intracellular signalling pathways: RAS/mitogen activated protein kinase (MAPK)/extracellular signal regulated kinase (ERK) or phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) (Figure 1). Abnormalities in the signalling pathway involving transforming growth factor beta (TGF- β) are also found in vascular malformations. Abnormal activation of the signalling pathway disrupts the proliferation, migration, adhesion, differentiation, and apoptosis of endothelial cells.
3
Eventually, these mutations induce dysmorphogenesis vasculature network and vascular malformation via alterations in angiogenesis and lymphangiogenesis.
14
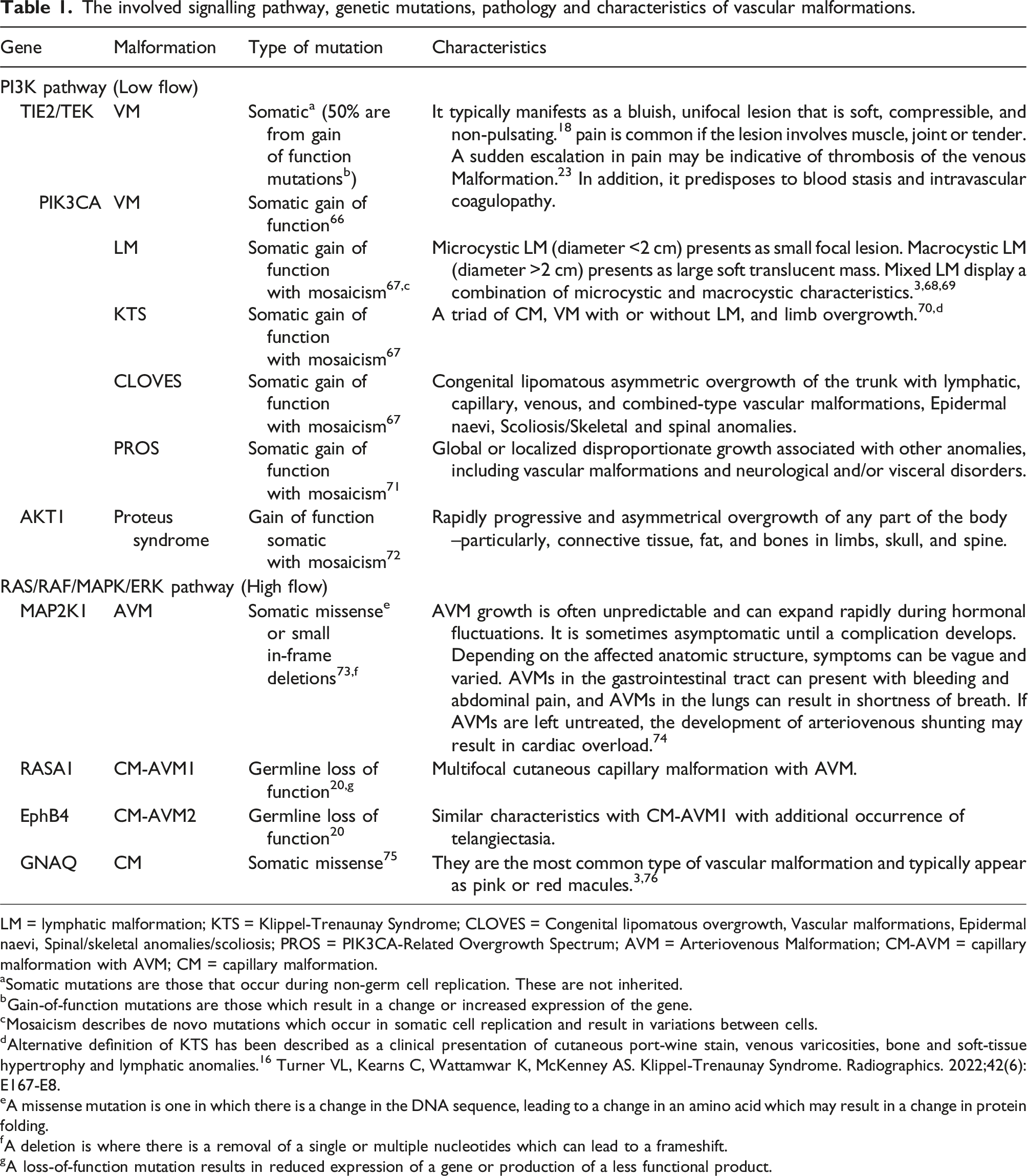
An overview of involved signalling pathway, genetic mutations, pathology and characteristics of vascular malformations is summarised in Table 1. Diagram illustrating the mutations and signalling pathways involved in vascular anomalies. ALK1, activin receptor-like kinase; AVM, arteriovenous malformations; CM, capillary malformation; EphB4, ephrin type-B receptor 4; ERK, extracellular signal regulated kinase; GNAQ, guanine nucleotide-binding protein G(q) subunit alpha; HHT, hereditary haemorrhagic telangiectasia; LM, lymphatic malformation; MEK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; PIK3, phosphatidylinositol-4,5-biphsophate 3-kinase catalytic subunit alpha; PTEN, phosphatase and tensin homolog; RASA1, RAS p21 protein activator 1; SMAD, homologues of the Drosphilia protein, mothers against decapentaplegic (MAD) and the Caenorhabditis elegans protein Sma; TIE2, TEK receptor kinase; VEGF, vascular endothelial growth factor; VM, venous malformation. The involved signalling pathway, genetic mutations, pathology and characteristics of vascular malformations. LM = lymphatic malformation; KTS = Klippel-Trenaunay Syndrome; CLOVES = Congenital lipomatous overgrowth, Vascular malformations, Epidermal naevi, Spinal/skeletal anomalies/scoliosis; PROS = PIK3CA-Related Overgrowth Spectrum; AVM = Arteriovenous Malformation; CM-AVM = capillary malformation with AVM; CM = capillary malformation. aSomatic mutations are those that occur during non-germ cell replication. These are not inherited. bGain-of-function mutations are those which result in a change or increased expression of the gene. cMosaicism describes de novo mutations which occur in somatic cell replication and result in variations between cells. dAlternative definition of KTS has been described as a clinical presentation of cutaneous port-wine stain, venous varicosities, bone and soft-tissue hypertrophy and lymphatic anomalies.
16
Turner VL, Kearns C, Wattamwar K, McKenney AS. Klippel-Trenaunay Syndrome. Radiographics. 2022;42(6):E167-E8. eA missense mutation is one in which there is a change in the DNA sequence, leading to a change in an amino acid which may result in a change in protein folding. fA deletion is where there is a removal of a single or multiple nucleotides which can lead to a frameshift. gA loss-of-function mutation results in reduced expression of a gene or production of a less functional product.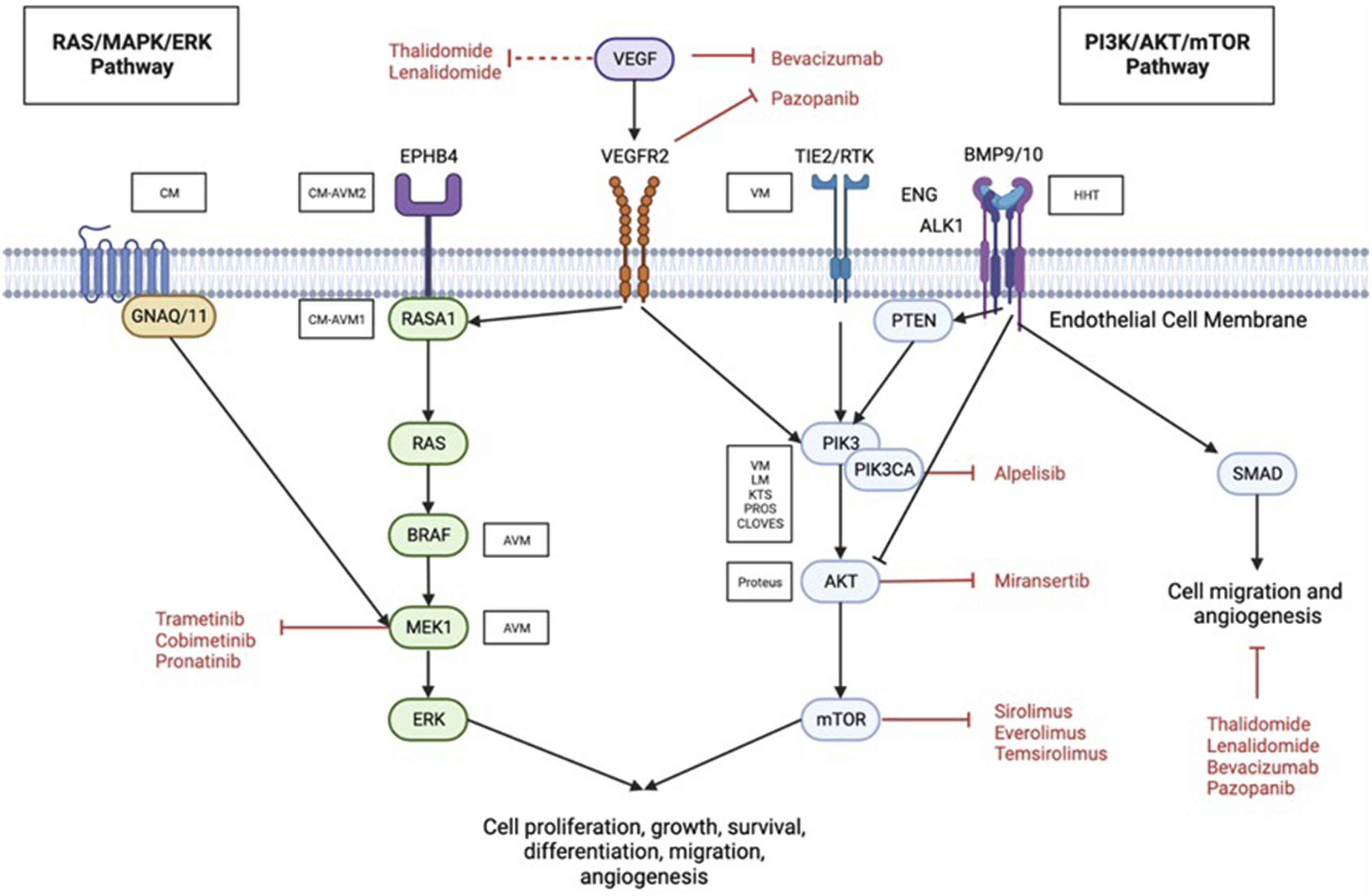
PI3K pathway
Important for cell cycle regulation, the PI3K pathway is also known as the “anti-apoptosis pathway.” The oncogene PIK3CA encodes the plasma membrane-associated lipid kinase PI3K. Ras, phosphatase and tensin homolog (PTEN), and VEGFR2 can activate PI3K in response to natural physiology or mutation-induced pathophysiology, among other factors. 17 This effect ultimately activates AKT and then mTOR; a serine/threonine protein kinase activates protein synthesis, which results in numerous cellular processes, including cell growth, proliferation, apoptosis, migration, migration, and angiogenesis. 13 Mutations in this pathway are associated with a variety of vascular anomalies. Low-flow vascular malformations especially those of venous and lymphatics vessels are specifically caused by various mutations in TIE2/TEK and oncogenic mutations of PIK3CA.18,19
PIK3CA mutations are also responsible for PIK3CA-related overgrowth spectrum (PROS), Klippel–Trenaunay Syndrome (KTS), Capillary Malformation of the Lower Lip Lymphatic Malformation of the Face/Neck Partial or Generalized Overgrowth Syndrome (CLAPO) and Congenital Lipomatous Overgrowth, Vascular malformations, Epidermal naevi and Scoliosis/skeletal/spinal anomalies (CLOVES). Gene AKT1 mutations cause Proteus syndrome.
RAS/MAPK/ERK pathway
The RAS/MAPK/ERK pathway plays a crucial role in the function of endothelial cells and is also known as the “proliferation pathway” (Figure 1). This pathway regulates the cell cycle and is responsible for cell proliferation and migration. Ras is a small GTP-binding protein that activates Raf serine/threonine kinase by dimerizing. Raf stimulates MAPL/ERK kinase (MEK), which in turn stimulates extracellular signal-regulated kinase (ERK). Gain of function mutations which result in increased expression or change in functioning of a gene or can lead to vascular malformations. ERK is a signal transduction protein that regulates cytosolic and nuclear molecules by transmitting mitogen signals.3,13 This pathway can also be activated pathologically by a number of mechanisms, including mutations in the VEFGR2, ephrin type-B receptor 4 (EphB4), and RAS p21 protein activator 1 (RASA1) protein-coding genes, as well as mutations in the guanine nucleotide-binding protein G(q) subunit alpha (GNAQ), GNA11, MAPK, and BRAF genes.
It has been established that a missense mutation, resulting in deletion of nucleotides in GNAQ causes CM, while a mutation in RASA1 causes CM-AVM1 (Figure 1). More recently, it was discovered that mutations in EphB4 that result in a loss of function are associated with the development of CM-AVM2. 20 MAP2K1 mutations are associated with the majority of sporadic AVMs.21,22 In addition, Hereditary Haemorrhagic Telangiectasia (HHT)-associated bleeding is often relevant to the presence of AVMs and also mutations in VEFG. 23 Therefore, agents that target the RAS/MEK/ERK pathway would be advantageous for treating the aforementioned vascular malformations especially the high flow lesions or AVMs.
Other involved singling pathways
TGF-β regulates endothelial cells by activating receptor-like kinase (ALK) 1 and ALK5. TGF-/ALK1 signalling stimulates endothelial cell migration, proliferation, and tube formation by activating SMAD1/5. TGF-/ALK5 signalling; on the other hand, inhibits angiogenesis by inhibiting endothelial cell proliferation, tube formation, and migration. (Figure 1)
HHT is caused by mutations in BMP9/10, ENG, Activin receptor-like kinase (ALK1), or SMAD4 in the TGF- β pathway, leading to an excess of VEGF. The VEGF signalling pathway is crucial for the regulation of angiogenesis, lymphangiogenesis, and vasculogenesis. ENG enhances BMP9/BMP10/Alk1 signalling, activating receptor-controlled SMAD. This inhibits endothelial cell migration and proliferation, maintaining the endothelium’s quiescent state. Patients with HHT may also develop CLOVES, Fibroadipose vascular anomalies (FAVA), KTS, lymphatic malformations, megalencephaly-capillary malformation (MCM), proteus syndrome, venous malformation, blue rubber bleb nevus syndrome (BRNBS) or PTEN Tumor hamartoma syndrome because of PI3K-AKT-mTOR pathway mutations. 24 HHT patients with RAS/Raf/MEK/ERK pathway mutations have extracranial AVMs, Kaposiform lymphangiomatosis (KLA), capillary malformation-AVMs and Sturge Weber syndrome.
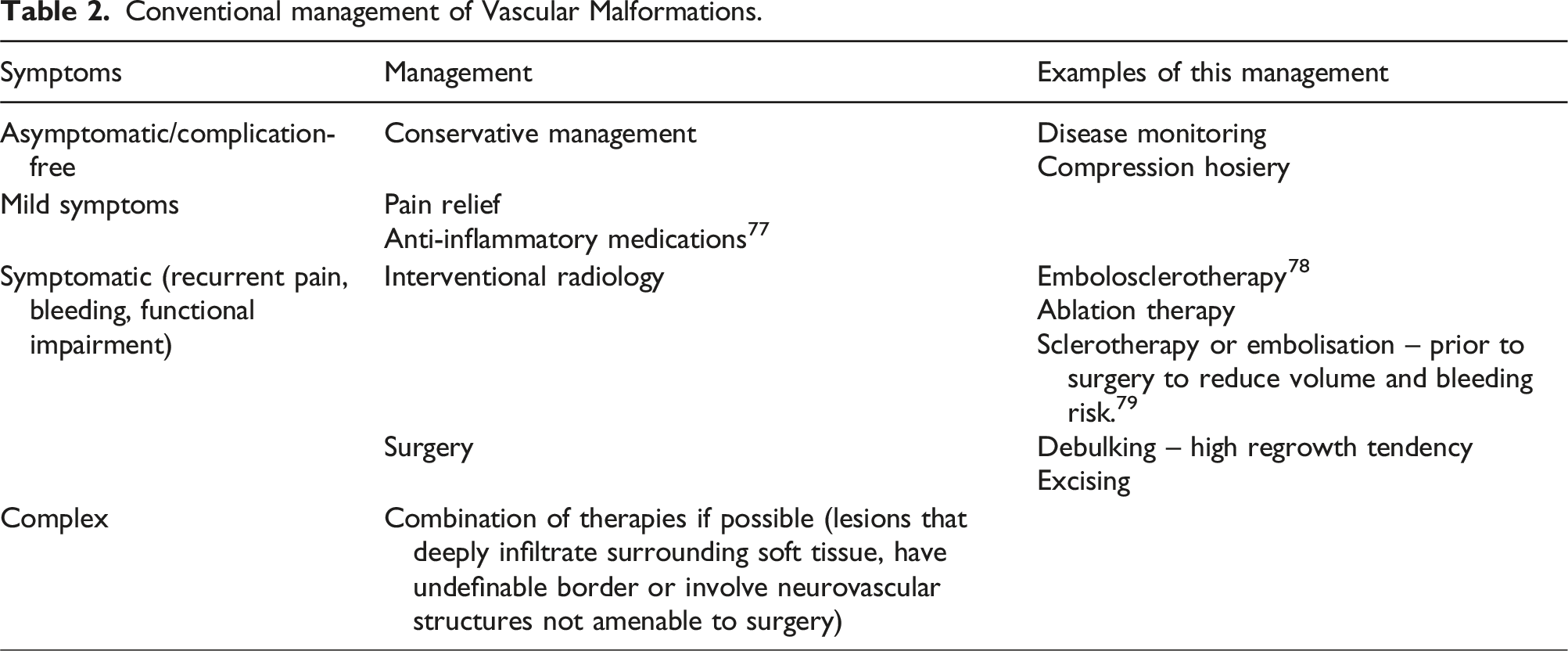
Conventional therapeutic management
Conventional management of Vascular Malformations.
Evolving pharmacological agents
Potential pharmacological agents in treating vascular malformations.
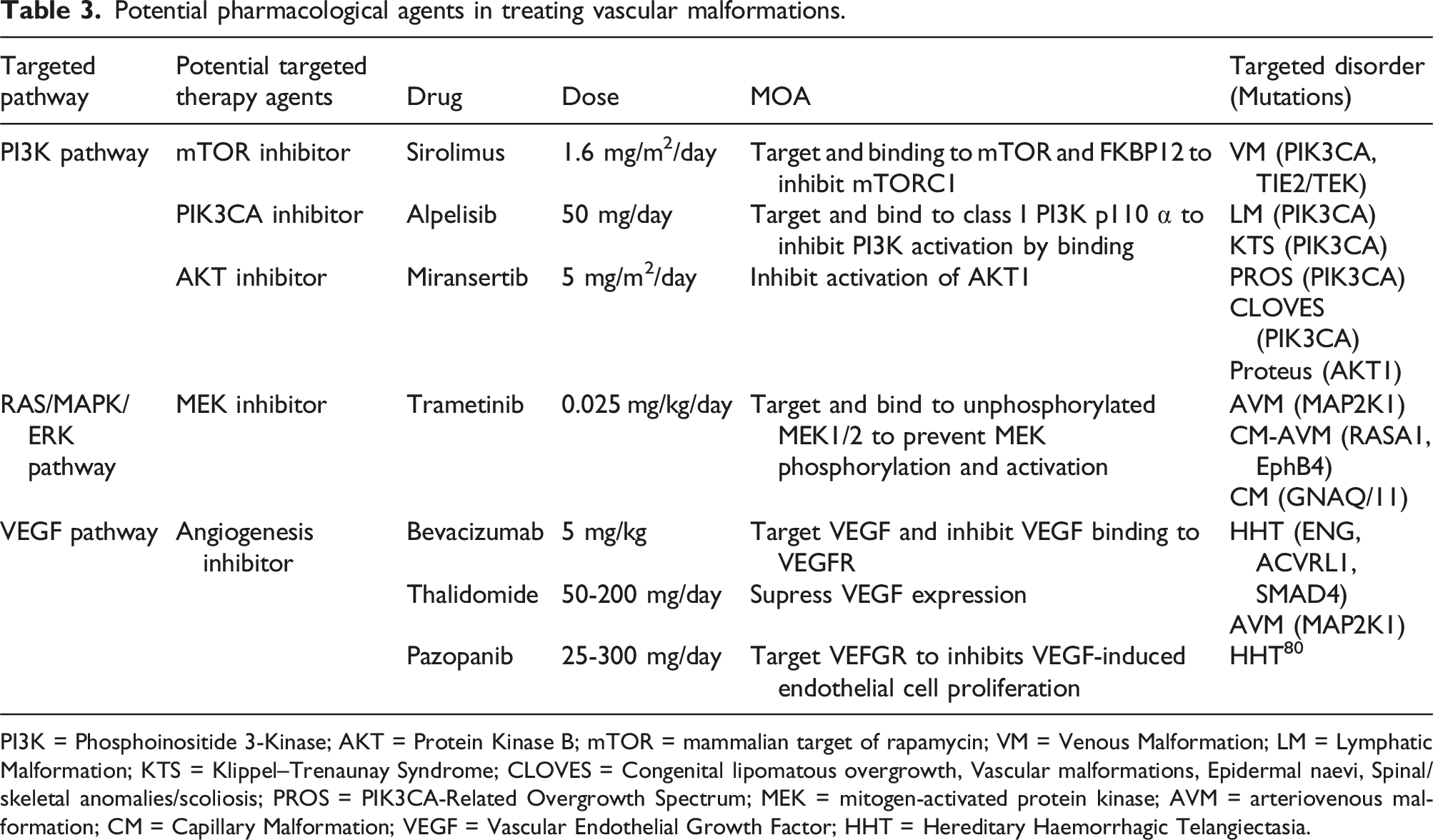
PI3K = Phosphoinositide 3-Kinase; AKT = Protein Kinase B; mTOR = mammalian target of rapamycin; VM = Venous Malformation; LM = Lymphatic Malformation; KTS = Klippel–Trenaunay Syndrome; CLOVES = Congenital lipomatous overgrowth, Vascular malformations, Epidermal naevi, Spinal/skeletal anomalies/scoliosis; PROS = PIK3CA-Related Overgrowth Spectrum; MEK = mitogen-activated protein kinase; AVM = arteriovenous malformation; CM = Capillary Malformation; VEGF = Vascular Endothelial Growth Factor; HHT = Hereditary Haemorrhagic Telangiectasia.
mTOR inhibitor (Sirolimus)
mTOR is a protein kinase that controls cellular proliferation, survival, and growth. The mTOR inhibitor inhibits downstream signalling and protein synthesis by preventing phosphorylation, reducing proliferation, and decreasing survival-related proteins.
Sirolimus, also known as Rapamycin, inhibits its mammalian target, mTOR, directly. It prevents the phosphorylation of Ribosomal protein S6 (S6RP) and 4E-BP1 by binding directly to its mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) (eukaryotic translation initiation factor 4E-binding protein 1). It then inhibits AKT activation and deregulates the PI3KCA/AKT/mTOR pathway. In the treatment of cancers and organ transplantation, Sirolimus is traditionally prescribed as an immunosuppressive, antiangiogenic, and cytostatic agent. It is also the first targeting agent being studied for the treatment of vascular malformations, and various studies and clinical trials have demonstrated its efficacy. 13
In a subsequent prospective clinical pilot study involving 6 patients with highly symptomatic VMs or combined syndromic VMs, sirolimus was shown to reduce pain, bleeding, lesion size, intravascular coagulopathy, functional and aesthetic impairment, and improve quality of life. 30 A study of seven patients with head and neck low flow vascular malformations found Sirolimus to be safe and effective. 31 Multiple phase 2 clinical trials also demonstrated the effectiveness, efficacy, safety in use, and tolerability of Sirolimus in treating adults and children with a variety of vascular malformations and tumours. 32 In the largest ongoing prospective multicentric phase 3 clinical trial enrolling 132 adult and paediatric patients with complex low-flow vascular malformations, including VMs, CMs, and LMs, that are resistant to standard treatment, Sirolimus was also reported to have a positive effect. 33 In the preliminary evaluation of the first 101 patients (70 cases of VM, 14 cases of LMs, 4 cases of CMs, 6 cases of KTS or CLOVES, 4 cases of GLA, 2 cases of GSD, and 1 case of PHTS), 87% demonstrated a reduction in pain and functional limitation, as well as an improvement in quality of life (NCT02638389).
Although Sirolimus is generally well tolerated, with oral mucositis, dyslipidaemia, leukopenia, gastrointestinal symptoms, and rash or eczema being the most frequently reported side effects, there have been reports that Sirolimus may increase the risk of infection as a complication. 34 In a prospective study involving 39 patients with PROS, it was reported that Sirolimus was ineffective and did not improve quality of life. 34 A study of Sirolimus treatment in patients with complex malformations resulted in two patients (3%) being removed from the study due to persistent nausea and lymphoedema which were not resolved with a dose reduction. 35 The most common Grade 2 and higher toxicities reported in this study were blood/bone marrow, gastrointestinal, infection, metabolic and pain related.
Although Sirolimus is highly selective for mTOR, they are effective at inhibiting mTORC2 and do not block all mTOR1 actions. 30 In addition, despite the promising effects of Sirolimus in VM and LM, there has been limited use for high-flow lesions or AVMs resulting in mixed results and there were few studies on Sirolimus’s effects on CM. 36
PI3KCA inhibitor (Alpelisib)
Several pharmacological PIK3CA inhibitors are currently being researched. Alpelisib is a PI3K p110 subunit-specific inhibitor. Class I PI3K p110, a catalytic subunit of PI3K, a lipid kinase that regulates cellular proliferation, survival, differentiation, and metabolism, is selectively inhibited. Because of its potent antitumor activity, it has been approved for use in oncology to treat breast cancer. 37
PIK3CA mutations are found in the majority of LM and 25–30% of VM. Alpelisib has been shown to effectively target downstream signalling of TIE2-mutated VMs, PIK3CA-mutated VMs, and MPIK3CA-mutated LMs.
Based on these encouraging results, a subsequent clinical study was conducted on six patients with severe PIK3CA-mutated LMs that were resistant to rapamycin, percutaneous sclerotherapies, and debulking surgery. Alpelisib was associated with improvements in each of the six patients, including the reduction of LMs, pain, and inflammatory flares. In this study, Alpelisib was generally well tolerated with the exception of a grade 1 mucositis and diarrhoea. 38 Alpelisib can induce hyperglycaemia so it is important to regularly monitor blood glucose levels to reduce the risk of hypoglycaemic episodes, for example with flash glucose monitoring. 39
In addition, PIK3CA mutations play a role in PROS, KTS, CLOVES, and CLAPO as well. 18 In a subsequent clinical study involving 19 patients with PROS, Alpelisib demonstrated improvement in disease symptoms in every patient without any reported adverse effects. 40 A case study also demonstrated the efficacy of Alpelisib in treating CLOVES. After 12 months of treatment, the following measurement revealed regression of adipose overgrowth and low flow vascular malformation with no other adverse effects. 41
AKT inhibitor (Miranseritib)
AKT is an essential serine-threonine protein kinase in the PI3K signalling pathway, which controls cell proliferation, survival, and motility. 42
Miransertib (ARQ092, MK-7075) is a novel, orally available, potent and specific AKT1 isoform-selective allosteric AKT inhibitor. 43 It suppresses AKT activity by inhibiting the membrane-bound active form of AKT, thereby preventing the activation of the inactive form of AKT. 44
Multiple clinical studies and case reports demonstrated that Miransertib could inhibit AKT signalling in Proteus syndromes with a favourable safety profile and long-lasting benefits in reducing Proteus symptoms, such as pain, and slowing the progression of overgrowth.45,46 More recently, a non-randomized phase 0/1 study in six individuals over the age of 12 with Proteus syndrome demonstrated a 50% reduction in phosphorylated AKT (pAKT) in affected tissues in five of six patients 4 months after starting a well-tolerated Miransertib treatment dose of 5 mg/m2/day, with effects including pain reduction and reduction in cerebriform connective tissue nevus. 47 Future efficacy trials investigating the direct effect of Miransertib on a human subject with AKT-related vascular malformation could benefit Vascular Malformation patients.
MEK inhibitor (Trametinib)
MEK proteins catalyse the phosphorylation of ERK1, which mediates cell proliferation, differentiation, and apoptosis.
Trametinib is an allosteric MEK inhibitor that is not ATP-competitive. It binds to unphosphorylated MEK1/2 to prevent the phosphorylation and activation of MEK by Raf. Trametinib was the first MEK inhibitor approved for the treatment of melanoma by the FDA. Recently, it has been studied as a potential treatment for vascular anomalies involving the RAS/MAPK/MEK pathway. In two off-label case studies of Trametinib for the treatment of somatic MAP2K1-mutated and KRAS-mutated AVM, both cases demonstrated decreased AVM blood flow and vessel calibre after 6 months of Trametinib treatment.21,48 Trametinib maintained the stability of leg hypertrophy and improved AVM shunting in a patient with CM-AVM and cardiac compromise, according to a second case study published in 2022. 22 Possible side effects of Trametinib include diarrhoea, fatigue, peripheral oedema, and cardiac dysfunction. Case reports of trametinib use have also found severe uveitis, ocular hypertension, intraocular inflammation, and skin reactions in patients on Trametinib.49–53 Reducing the dosage or administering it for a shorter period of time has been suggested to mitigate these side effects. 54 Currently, two phase 2 clinical trials have started recruitment which are evaluating the safety and efficacy of Trametinib in patients with extracranial AVMs and AVMs resistant to standard treatment (NCT04258046 and EndraCT 2019-003573-26 respectively).
After the approval of Trametinib, other MEK inhibitors were being investigated. Cobimetinib is the second MEK inhibitor approved for the treatment of malignant melanoma. It is a highly potent and selective MEK inhibitor that has demonstrated in vivo efficacy in BRAF and KRAS-mutated cells in xenograft models. Currently, a phase 2 open-label clinical trial of Cobimetinib for extracranial AVMs is being conducted (NCT05125471). Ponatinib is yet another inhibitor of MEK. However, clinical research on the effect of Pronatinib on vascular malformation is limited.
Rigorous phase 2/3 randomised control trials are still needed to be more definitive in describing side effects and outcomes.
Antiangiogenic medications
VEGF is a key regulator of vasculogenesis and angiogenesis, acting as a highly specific and potent mitogen of vascular endothelial cells, promoting endothelial cell survival and migration, and increasing microvascular permeability.55,56
Increased expression has been demonstrated in the endothelium of AVM lesions57–60 and increased circulating VEGF levels in AVM patients compared to healthy controls. 61
Thalidomide is an antiangiogenic agent that inhibits VEGF expression.
In 2022, a prospective observational study involving 18 patients with severe symptomatic cranial (13 patients) and limb (5 patients) AVMs demonstrated that Thalidomide managed chronic pain, bleeding, and ulceration of extensive AVMs that are resistant to standard therapy. 62 By inhibiting TNF-alpha and nitric oxide, thalidomide also has anti-inflammatory and immunomodulatory effects. The multiple functions of thalidomide may account for its efficacy in treating AVM in this study, particularly when combined with embolisation. However, the use of Thalidomide has been restricted due to its side effects and potential complications, which includes teratogenicity.2,62,63 Therefore, improved thalidomide analogues such as Lenalidomide, with fewer side effects were developed. Despite positive results in preclinical studies, human subject studies and clinical studies comparing between Thalidomide and Lenalidomide are limited.
Bevacizumab is an anti-VEGF monoclonal antibody directed against VEGF-A. 64 It has a potent anti-angiogenic effect in the treatment of neo-angiogenesis. It functions by binding selectively to circulating VEGF to inhibit VEGF binding to its cell surface receptors, thereby reducing angiogenesis. It is approved and recommended for the treatment of advanced solid cancers. Due to its antiangiogenic effect, it is regarded as a possible treatment for AVM and HHT. Multiple studies including phase 1 and phase 2 studies have demonstrated that Bevacizumab is safe and effective in the treatment of bleeding associated with intestinal, nasal, pulmonary, and hepatic AVMs in patients with HHT. 64 A pilot study did not find adverse effects associated with Bevacizumab in two patients who underwent intravenous infusions of 5 mg/kg every 2 weeks for 12 weeks. 65 However, research on the efficacy and safety of Bevacizumab in the treatment of isolated AVM is limited.
Pazopanib is a small-molecule inhibitor of VEGF receptor tyrosine kinase. In a recent observational study involving 13 patients with severe HHT-associated bleeding and red cell transfusion-dependent anaemia, low-dose oral Pazopanib demonstrated significant haemostatic efficacy in managing the condition. However, the use of pazopanib is still in its infancy, and additional research is required to determine its efficacy against HHT and potentially other vascular malformations involving the relevant signalling pathway. 24
Conclusion and future direction
The recent improvement in the understanding of disease classification and terminology has made it possible to recommend accurate diagnoses and treatments. In addition, a better understanding of the genetic and molecular mechanisms underlying vascular malformation has facilitated the development of targeted therapies and increased our knowledge of its pathophysiology. Historically, surgical or minimally invasive techniques were used to treat vascular malformations. Currently, targeted therapeutic research and drug repurposing are being conducted based on the underlying cellular pathways involved and innovative pharmacological treatment has the potential to induce remission of vascular malformation and substantially improve the quality of life of the patients. Genetic testing of affected tissue remains a diagnostic challenge for certain vascular anomalies where tissue biopsy is difficult or carries a high risk. Additionally, the optimal dosage, blood levels, and treatment duration must be determined. However, as more genetic information is gathered and added to the current genetic database, precision medicine can be enhanced which will ultimately enable clinicians to personalise management strategies for their individual patient with vascular malformations. As vascular malformations typically develop after birth and may necessitate long-term treatment, the long and short term safety profile of therapeutic agents must be investigated to ensure safe. With future research on the pharmacological therapy of vascular malformation, medical treatment is likely to be an effective addition to the therapeutic options for these often difficult-to-treat entities. It can not only improve patient outcomes, but also potentially reduce the psycho-social burden for managing this chronic disease.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: CP received funding from the Royal Free Charity, London, United Kingdom. This study was supported by researchers (CSL) at the National Institute for Health Research University College London Hospitals Biomedical Research Centre. The Royal Free Vascular Anomalies also received funding from the Butterfly AVM Charity.
Contributorship
CP, RL an RN researched literature, conceived the study and wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and all authors approved the final version of the manuscript.