
Abstract
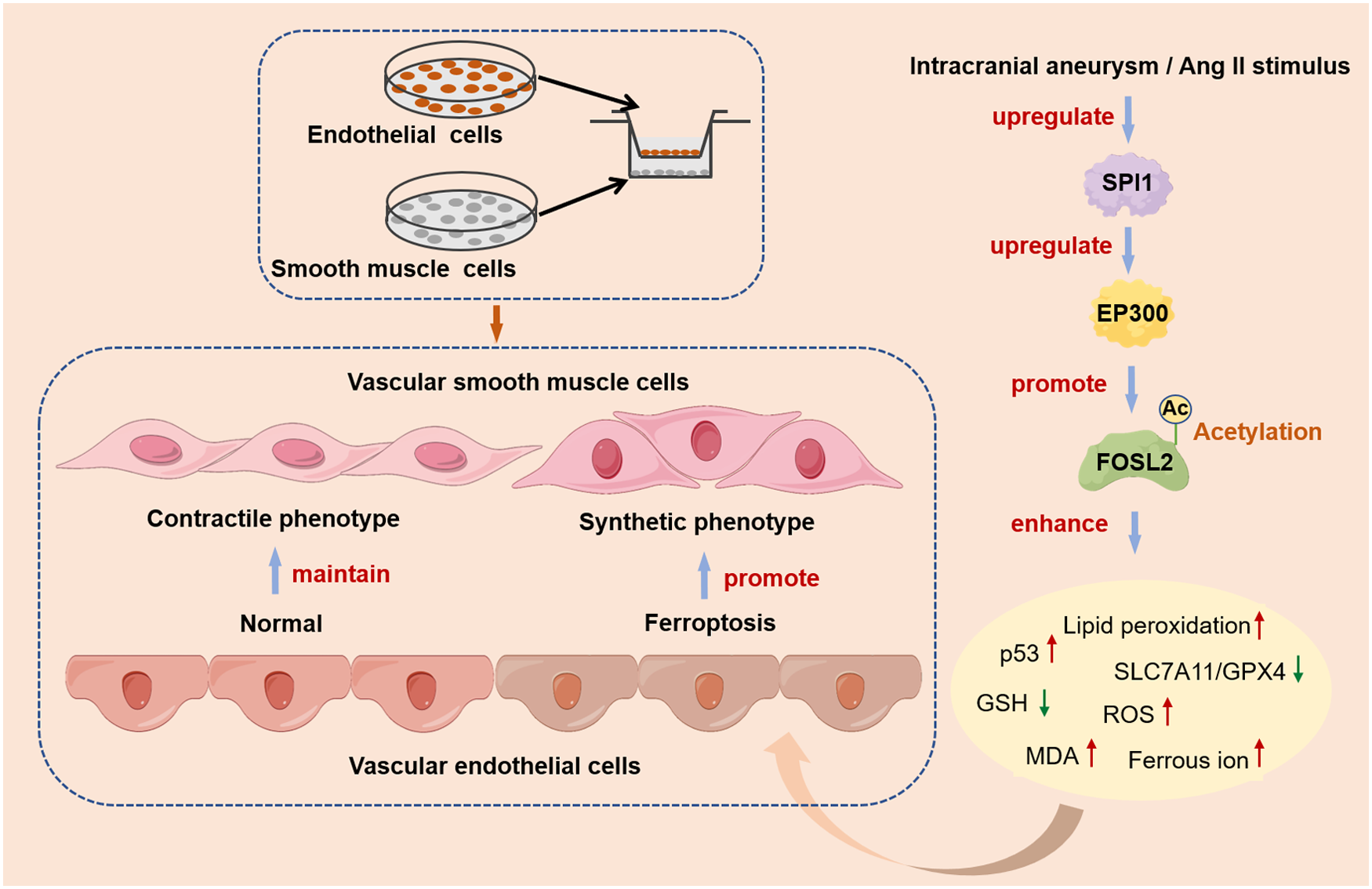
This study investigates the role of SPI1 in the pathogenesis of intracranial aneurysm (IA). Results confirmed that SPI1 was significantly upregulated in both an in vivo mouse IA model and an in vitro model of angiotensin II-stimulated human cerebral microvascular endothelial cells. Functional studies demonstrated that SPI1 overexpression promoted vascular endothelial cell ferroptosis, as evidenced by dysregulation of key markers (p53, GPX4, SLC7A11, ACSL4, TFRC), elevated lipid peroxidation, and mitochondrial damage, whereas SPI1 knockdown attenuated these effects. Mechanistically, SPI1 upregulated the histone acetyltransferase EP300, which subsequently mediated the acetylation of FOSL2. Acetylated FOSL2 enhanced the transcriptional activity of the p53 promoter, thereby driving the execution of ferroptosis. In a co-culture system, SPI1-induced vascular endothelial ferroptosis facilitated the phenotypic switching of vascular smooth muscle cells towards a synthetic phenotype. Crucially, administration of the SPI1 inhibitor DB2313 effectively suppressed the SPI1/EP300/FOSL2 axis, inhibited vascular endothelial ferroptosis, mitigated vascular smooth muscle cell phenotypic switching, and alleviated IA progression in mice. Our findings revealed a novel SPI1/EP300/FOSL2 acetylation axis that promoted vascular endothelial ferroptosis and vascular smooth muscle cell dysfunction in IA, identifying SPI1 as a promising therapeutic target for the condition.
Keywords
Get full access to this article
View all access options for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.