
Abstract
Polyuria and polydipsia are the characteristics of congenital nephrogenic diabetes insipidus (CNDI). Approximately 90% of all patients with CNDI have X-linked hereditary disease, which is due to a mutation of the arginine vasopressin receptor 2 (AVPR2) gene. This case report describes a 54-year-old male with polyuria and polydipsia and several male members of his pedigree who had the same symptoms. The proband was diagnosed with diabetes insipidus using a water-deprivation and arginine vasopressin stimulation test. Genomic DNA from the patient and his family members was extracted and the AVPR2 gene was sequenced. A novel missense mutation of a cytosine to guanine transition at position 972 (c.972C > G) was found, which resulted in the substitution of isoleucine for methionine at amino acid position 324 (p.I324M) in the seventh transmembrane domain of the protein. The proband’s mother and daughter were heterozygous for this mutation. The novel mutation of the AVPR2 gene further broadens the phenotypic spectrum of the AVPR2 gene.
Introduction
Congenital nephrogenic diabetes insipidus (CNDI) is a relatively rare hereditary disease, which is commonly diagnosed by characteristic symptoms, such as polyuria, polydipsia, fever with unknown aetiology, convulsions, vomiting and constipation in early infancy. CNDI is caused by a mutation of the arginine vasopressin receptor 2 (AVPR2) gene or the aquaporin 2 gene (AQP2) gene.1,2 Mutation of the AVPR2 gene leads to X-linked NDI (X-NDI), 3 which accounts for 90% of all diagnosed CNDI cases. 4
The AVPR2 gene was first identified in 1992. 1 AVPR2 maps to chromosome X (Xq28), and thus CNDI is transmitted in an X-linked recessive mode (OMIM 304800); 5 males with the mutated gene are symptomatic, whereas heterozygous females are usually asymptomatic. The AVPR2 protein contains seven membrane-spanning helices. 1 Upon binding with arginine vasopressin (AVP), activation of the receptor is initiated, and allosteric structural rearrangements occur. 6 AVP binds to the AVPR2 within the transmembrane helices II–IV.7,8 Mutations in the AVPR2 gene can affect the binding between AVP and AVPR2 or signal transduction, which prevents the renal tubules from concentrating urine and produces the symptoms of polyuria and polydipsia.
The current report presents a case of a Chinese pedigree with CNDI that has a novel missense mutation detected through sequence analysis of the AVPR2 gene.
Case report
A 54-year-old man, presenting with left limb weakness for 1 week, was admitted to the Department of Neurology, Tianjin Medical University General Hospital, Tianjin, China, on 24 October 2014 with the preliminary diagnosis of cerebral infarction. Dot diffusion-weighted magnetic resonance imaging (MRI) of the head showed high signal intensity, and a softened lesion was found in the right side of the basal ganglia region. The patient had complained of polydipsia and polyuria since birth. He drank 10–15 l of water per day, and his urine output was large as well with nocturia for 5–6 times per night. The patient had no recent history of using renal injury agents. His family history showed his grandmother and grandfather had a consanguineous marriage (they were first cousins). Seven individuals in this pedigree have the same symptoms of polydipsia and polyuria. His younger brother died from dehydration when he was an infant. Because of his seniority and siblings sharing the same symptoms, he had not paid attention to the disease, and received no diagnosis or treatment.
Water-deprivation and arginine vasopressin stimulation test on a 54-year-old man with suspected diabetes insipidus.
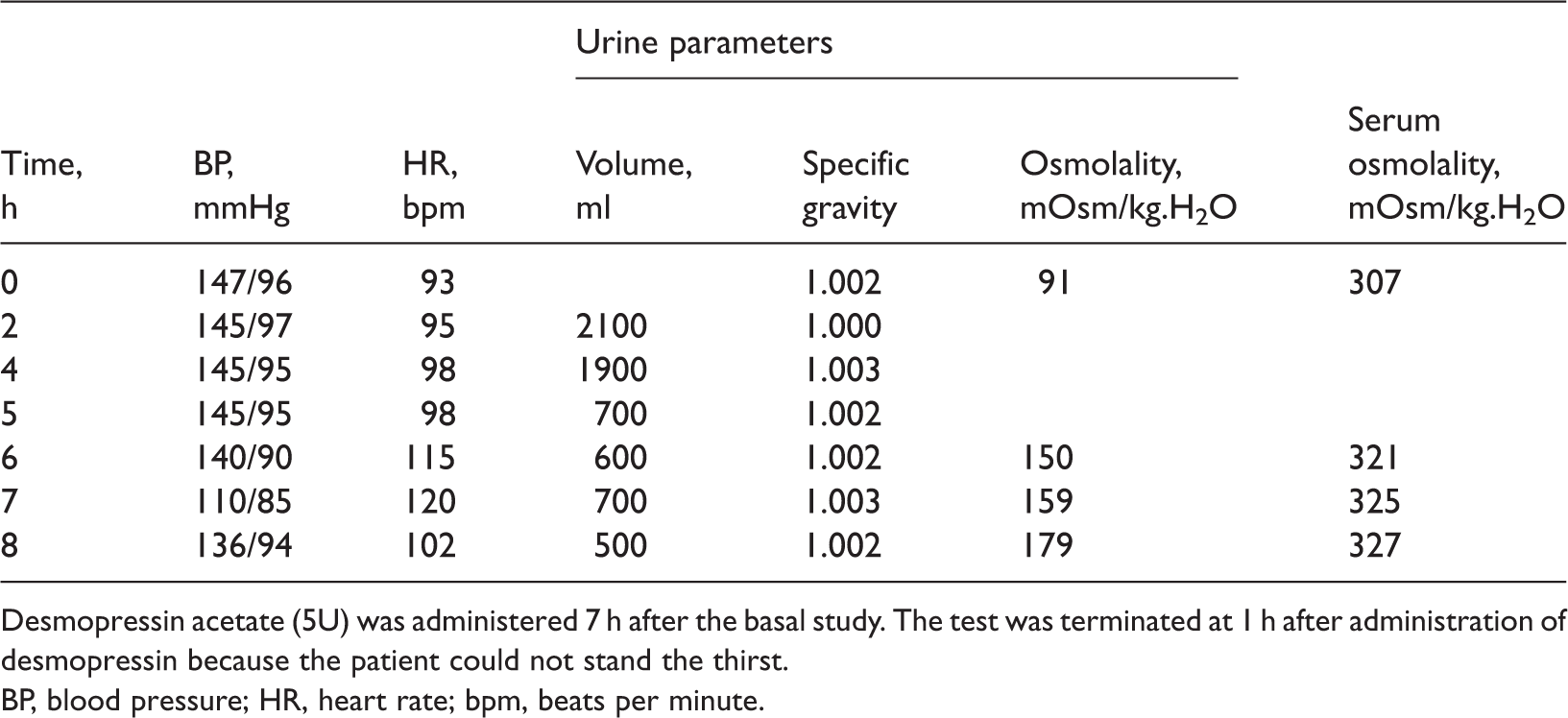
Desmopressin acetate (5U) was administered 7 h after the basal study. The test was terminated at 1 h after administration of desmopressin because the patient could not stand the thirst.
BP, blood pressure; HR, heart rate; bpm, beats per minute.
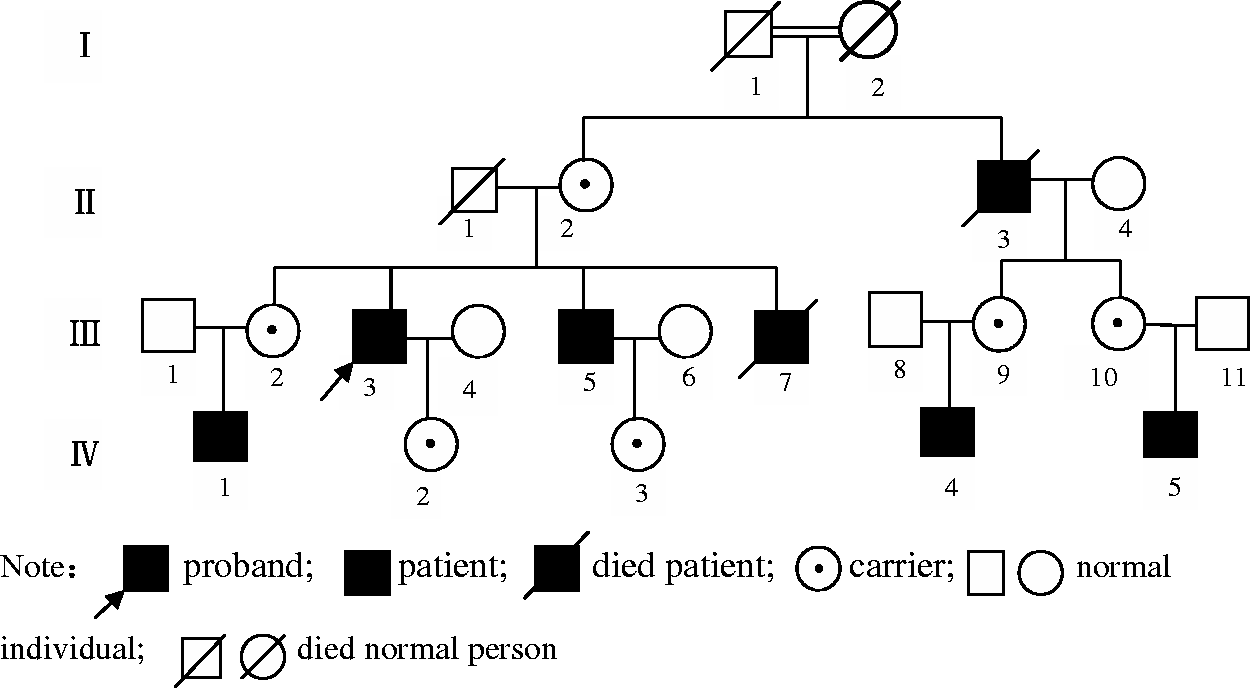
The pedigree map of the proband who was a 54-year-old man with suspected diabetes insipidus.
Polymerase chain reaction primers used to amplify four exons of the arginine vasopressin receptor 2 gene.

Analysis of the AVPR2 gene sequences of the proband, his brother and nephew revealed a novel missense mutation at coding position 972 (c.972C > G) (Figure 2). This mutation resulted in a change of the 324th amino acid from isoleucine into methionine (p.I324M). His mother, sister, niece and daughter were detected as heterozygotes for the same mutation. His wife was not identified as having an AVPR2 mutation. The male offspring of the proband’s female cousins refused to undertake genetic testing, so were not analysed.
DNA sequencing results for the 54-year-old man with suspected diabetes insipidus and seven first- and second-degree relatives. Arrows represent the mutation site. The colour version of this figure is available at: http://imr.sagepub.com.
To assess the severity of the mutation in silico, the mutation was analysed using Polymorphism Phenotyping v2,9,10 which predicts the possible impact of an amino acid substitution on the structure and function of a human protein using physical and evolutionary comparative considerations. The software predicted the mutation’s effect with a score of 0.998 (out of 1.0), sensitivity of 0.18 and specificity of 0.98, therefore making p.I324M a probable pathogenic mutation.
The study was conducted in accordance with the principles of good clinical practice and the Declaration of Helsinki and was approved by the Ethics Committee of Tianjin Medical University General Hospital. Written informed consent was obtained from all participants for publication of this case report and the accompanying images.
Discussion
In this present study, a pedigree from a consanguineous marriage had several individuals who had experienced polydipsia and polyuria since their infancy. Based on family histories, clinical symptoms (polyuria and polydipsia), laboratory tests (serum sodium, urine specific gravity, urine and serum osmolalities), and the response to a water-deprivation and arginine vasopressin stimulation test, the proband was diagnosed with NDI, and an X-linked recessive CNDI pedigree was established. Further AVPR2 gene sequence analysis identified a novel missense mutation at coding position 972 (c.972C > G), which has not been reported before.
Most patients with CNDI are usually identified in infancy due to their persistent polyuria, severe dehydration, frequent fever, delayed growth, a condition that was first described by McIlraith in 1892. New born infants with NDI often suffer from dehydration, hypernatraemia and seizures, and if it is not diagnosed in a timely manner, those symptoms can be life threatening. In the present study, the proband had experienced a long-term fever during infancy for which an explanation was never found. His younger brother died from dehydration and because several individuals with the same symptoms existed in his family, he did not pay close attention to the disease. The females of the proband’s pedigree did not have the symptoms of polydipsia and polyuria. DNA sequence analysis of the proband’s mother, sister, niece and daughter showed that they were heterozygous carriers of the mutation. Due to the presence of a normal allele in these females, they had no symptoms of polydipsia or polyuria. All of the patients in this pedigree with symptoms of polydipsia or polyuria were men, which can be explained by the fact that males have only one X chromosome. Female heterozygous carriers with NDI symptoms can be explained by an extremely skewed inactivation of the normal allele of the X chromosome, 11 but the prevalence of symptomatic female heterozygotes remains unknown.
To date, according to the Human Gene Mutation Database, 12 about 258 mutations in the AVPR2 gene causing NDI have been identified, including missense mutations, nonsense mutations, splicing mutations and frameshift mutations leading to a premature stop codon.13,14 Missense mutations are the most common mutated type. 15 An AVPR2 mutation can cause abnormal protein folding after mRNA translation. 15 In vitro experiments have established that most AVPR2 mutations lead to the encoded receptors being trapped inside the endoplasmic reticulum. 16 A few copies of the mutated receptors can reach the cell surface, but the AVP binding and the Gs protein coupling are impaired, which results in failure to trigger the intracellular cascade of adenylate cyclase effectively. 17 The novel mutation reported by this present study maps to the seventh transmembrane domain of the protein.
One limitation of this present study was the lack of functional studies of the mutated gene. Missense mutations in this region (Trp323, Ger329) have been reported to cause NDI in European populations.18,19 These studies highlighted the sequence conservation and functional importance of this region.18,19
In conclusion, this present case report describes a CNDI pedigree with an AVPR2 gene I324M missense mutation. To the best of our knowledge, this is the first report of this mutation in patients with CNDI and broadens the phenotypic spectrum of AVPR2 mutations. Recently, new drugs were found to treat CNDI,2,20 so identifying AVPR2 gene mutations may be helpful for specific treatment.
Footnotes
Acknowledgments
We are very grateful to the family for their contribution and the KingMed Center for Clinical Laboratory Company Limited, Guangzhou, China for gene sequencing.
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.