
Abstract
Objective
Arthritis, including rheumatoid arthritis, gonarthrosis, and gouty arthritis, poses a substantial public health challenge. Regulatory T cells are critical for immune homeostasis, and their dysfunction contributes to arthritis pathogenesis; however, the specific gene regulatory programs causally influencing disease susceptibility remain elusive. We aimed to identify regulatory T cell–specific therapeutic targets using single-cell cis-expression quantitative trait locus and Mendelian randomization.
Methods
We integrated single-cell regulatory T cell cis-expression quantitative trait locus data with genome-wide association study results from FinnGen R12 (16,314 rheumatoid arthritis, 61,356 gonarthrosis, and 12,342 gouty arthritis cases). Two-sample Mendelian randomization was employed to infer causality, using the inverse-variance weighted method, with rigorous sensitivity analyses conducted to ensure robustness.
Results
The analysis revealed distinct causal associations. In rheumatoid arthritis, upregulation of DexD/H-box helicase 60 like (DDX60L) significantly increased the disease risk (odds ratio = 2.10), whereas OAS1 expression served as a protective factor (odds ratio = 0.65). For gonarthrosis, OAS1 again demonstrated a protective effect (odds ratio = 0.897), whereas janus kinase and microtubule interacting protein 2 (JAKMIP2) was identified as a modest risk factor (odds ratio = 1.019). Conversely, no significant regulatory T cell–specific causal signals were observed for gouty arthritis.
Conclusions
OAS1 is a cross-disease regulatory T cell protector, whereas DDX60L and JAKMIP2 are disease-specific risk amplifiers. Engineering high-OAS1/low-DDX60L/low-JAKMIP2 regulatory T cells offers a genetically grounded blueprint for rheumatoid arthritis and gonarthrosis but not for gouty arthritis.
Keywords
Introduction
Arthritis encompasses a spectrum of debilitating musculoskeletal disorders characterized by joint inflammation and structural degradation, among which rheumatoid arthritis (RA), gonarthrosis (GA), and gouty arthritis (GOA) represent major public health challenges with distinct yet overlapping pathological features.1–3 RA, an autoimmune disease affecting approximately 1% of the global population, is characterized by synovial inflammation and progressive joint destruction4–6; GA, the most prevalent degenerative joint disease, primarily impacts the knee joint in middle-aged and older individuals, leading to cartilage loss and functional impairment;7–9 GOA, triggered by monosodium urate crystal deposition, is associated with acute inflammatory flares and long-term joint damage.10–11 Despite advances in therapeutic strategies, including disease-modifying antirheumatic drugs, nonsteroidal anti-inflammatory drugs, and targeted biologics, many patients exhibit inadequate response, adverse effects, or disease progression, thereby highlighting an urgent need for novel, mechanism-driven therapeutic approaches.12,13
Central to the maintenance of immune homeostasis and self-tolerance are regulatory T cells (Tregs), a specialized cluster of differentiation (CD) CD4+ T cell subset characterized by the expression of the transcription factor FoxP3.14–16 Tregs play a critical role in suppressing aberrant immune responses through various mechanisms, including the secretion of anti-inflammatory cytokines and modulation of antigen-presenting cell function.17,18 In the context of arthritis, functional defects or numerical deficiencies in Treg populations are frequently observed, correlating with disease severity and progression.19,20 Consequently, Treg-based immunotherapies, such as the infusion of ex vivoexpanded Tregs or the engineering of chimeric antigen receptor (CAR)-Tregs, have emerged as promising avenues for restoring immune tolerance in autoimmune diseases, transplantation, and graft-versus-host disease.21,22 These approaches aim to enhance the number, specificity, and suppressive function of Tregs, thereby promoting antigen-specific immune regulation without broad immunosuppression.21–23
However, the clinical translation of these therapies is hindered by the incomplete understanding of the specific gene regulatory programs within Tregs that causally influence arthritis pathogenesis. Identifying precise molecular targets that can modulate Treg function without inducing general immunosuppression is paramount for optimizing these cellular therapies.
Disentangling the causal relationship between Treg-specific gene expression and arthritis risk from observational data is challenging due to the presence of confounding factors and reverse causation. Mendelian randomization (MR) offers a robust solution by utilizing genetic variants as instrumental variables (IVs) to infer causality, thereby minimizing bias from environmental confounders.24,25 Furthermore, the integration of MR with single-cell expression quantitative trait loci (eQTL) (sc-eQTL) data allows for an unprecedented resolution of genetic regulation. Unlike bulk eQTL analyses, sc-eQTLs can delineate cell type–specific regulatory mechanisms that are often masked in tissue-level averages.26–28 Given that Tregs possess unique transcriptional landscapes and epigenetic modifications, cis-eQTLs identified in Tregs likely reveal distinct regulatory pathways specific to immune tolerance.27,28 This specificity is crucial, as genetic variants that regulate gene expression in Tregs may not exert the same effects in other immune or stromal cells.
In this study, we aimed to bridge the gap between immunogenetics and therapeutic bioengineering by conducting an integrated analysis of single-cell Treg cis-eQTLs and two-sample MR. We systematically evaluated the causal effects of genetically predicted Treg-specific gene expression on the susceptibility to RA, GA, and GOA. By leveraging large-scale genome-wide association study (GWAS) summary statistics and high-resolution sc-eQTL datasets, we sought to identify key Treg-restricted genes—such as potential risk factors or protective agents—that drive arthritis pathogenesis. The findings from this research are intended to provide a genetic blueprint for the rational design of next-generation Treg therapies, offering insights into how to selectively enhance protective pathways (e.g. upregulating beneficial genes such as OAS1) or inhibit detrimental ones (e.g. modulating risk genes such as, DexD/H-box helicase 60 like (DDX60L) or janus kinase and microtubule interacting protein 2 (JAKMIP2)), ultimately advancing the field toward more effective and personalized treatments for arthritis.
Methods
Study design and ethics
We executed this research adhering to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE)–MR guidelines to ensure methodological strictness. 29 To facilitate sound causal deduction, the study satisfied three fundamental tenets of MR: (a) first, genetic variants utilized as IVs must show a strong connection to the exposure; (b) second, these variants ought to be free from confounding elements; and (c) third, their effect on the outcome is mediated solely via the exposure of interest. Our methodology depended entirely on summary-level statistics available from public GWAS databases, which meant that individual-level data were not utilized, and no supplementary ethical permissions were needed. Prior investigations provided the necessary documentation confirming that the original GWAS datasets secured participant written informed consent and had received clearance from their respective ethics committees.
Exposure and instrument selection
For the establishment of reliable IVs in our MR analyses, we obtained cis-eQTL information specific to single-cell Tregs from the single-cell quantitative trait loci (scQTLs) database (http://bioinfo.szbl.ac.cn/scQTLbase/Home/), 30 which is a comprehensive sc-eQTL database that integrates eQTL data from multiple single-cell RNA–sequencing studies across diverse cell types and contexts. 30 We implemented a comprehensive, multistage screening protocol to guarantee both potency and credibility of our instruments. Initially, we identified conditionally autonomous cis-eQTLs demonstrating genome-wide statistical significance (p < 5.0 × 10−8), thereby mitigating the risk of spurious findings. Subsequently, we conducted linkage disequilibrium (LD) aggregation procedures utilizing the European population reference data from the 1000 Genomes Project, employing stringent parameters, including an LD cutoff of r2 <0.001 within a genomic interval of 10 megabases to secure variant autonomy. 31 Following this, we aligned the datasets corresponding to exposures and outcomes, discarding single nucleotide polymorphisms (SNPs) with ambiguous strand orientation, except where allele frequency data enabled definitive orientation. Furthermore, to circumvent weak-instrument distortion, we computed F-statistics for individual SNPs and eliminated variants displaying F-values <10.32,33 This benchmark serves to constrain the potential distortion of MR-derived effect estimates to roughly 10% relative to observational associations.34,35 Through the implementation of these rigorous inclusion criteria, we preserved exclusively robust and autonomous genetic instruments, thereby attenuating bias in our subsequent MR evaluations. Comprehensive details regarding the single-cell Treg cis-eQTLs incorporated in this investigation are documented in Table 1. Genes that did not reach nominal statistical significance (p < 0.05) in the MR analysis were excluded from the detailed results narrative to maintain focus on the primary causal targets; however, all screened instruments are documented in Table 1.
Treg-specific cis-eQTLs for Mendelian randomization analysis.
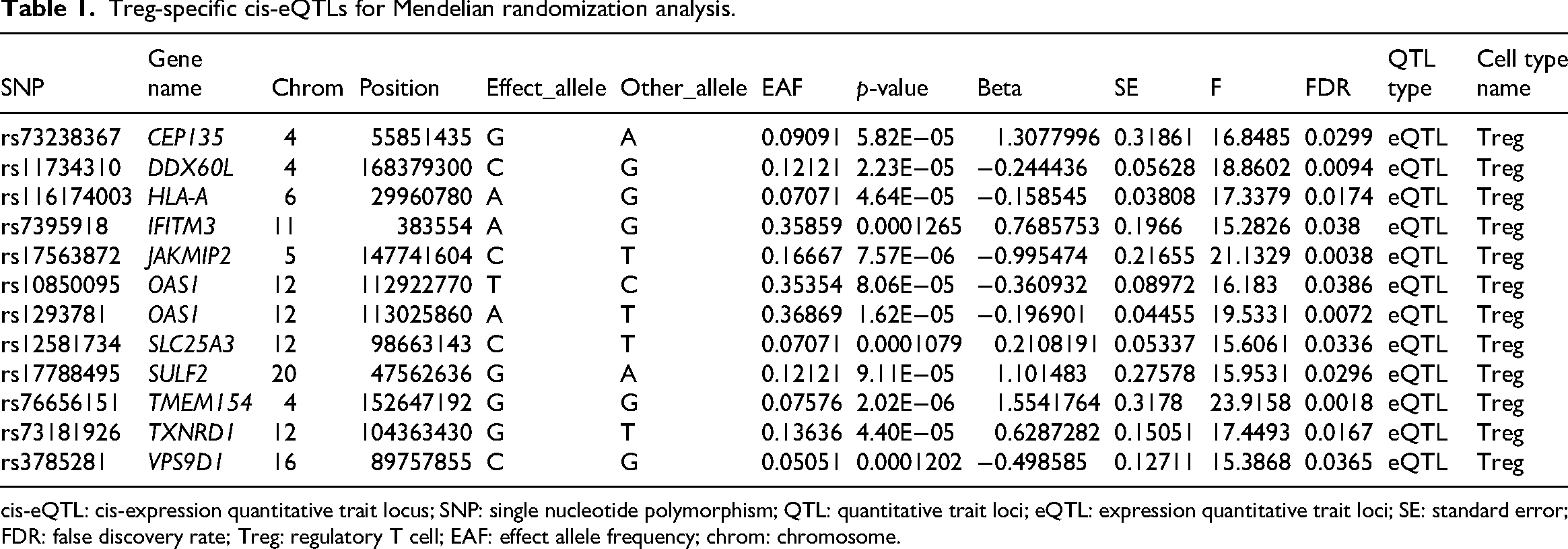
cis-eQTL: cis-expression quantitative trait locus; SNP: single nucleotide polymorphism; QTL: quantitative trait loci; eQTL: expression quantitative trait loci; SE: standard error; FDR: false discovery rate; Treg: regulatory T cell; EAF: effect allele frequency; chrom: chromosome.
Outcome data
Summary-level data for RA were obtained from the FinnGen consortium release R12 (data freeze 12, DF12; genome build GRCh38; public release version 1, 14 October 2025), comprising 16,314 cases alongside 315,115 controls. Similarly, the dataset for GA was retrieved from the FinnGen release, consisting of 61,356 patients and 315,115 healthy individuals. With respect to GOA, we utilized FinnGen-derived statistics, which included 12,342 patients and 315,115 controls. A complete breakdown of the utilized GWAS datasets has been detailed in Table 2.
Data sources for Mendelian randomization analysis.
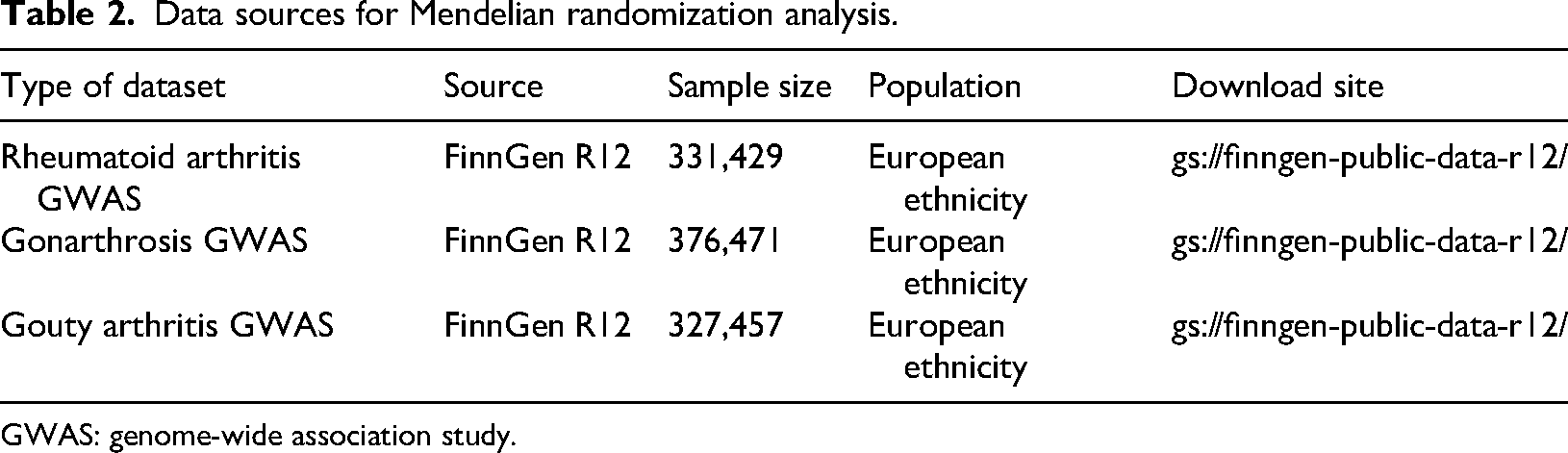
GWAS: genome-wide association study.
Sc-eQTL–MR analysis
To evaluate the causal influence of Treg-specific gene expression on conditions such as RA, GA, and GOA, we performed MR analysis utilizing the ‘TwoSampleMR’ R package software (version 0.6.29). We employed the Wald ratio technique for genes characterized by a single suitable instrumental SNP. Conversely, for genes associated with multiple independent SNPs, we utilized the inverse-variance weighted (IVW) method to calculate combined causal estimates. 36 Recognizing the necessity to adjust for multiple comparisons given the vast quantity of Treg-specific genes, we implemented a false discovery rate (FDR) correction tailored to the number of genes tested for each specific arthritis type. These methods allowed for the reliable determination of the potential causal roles played by Treg-specific gene regulation across these different arthritic conditions.
Sensitivity testing
For those genes identified with more than two independent IVs, we deployed supplementary MR methodologies—specifically MR-Egger, weighted median, simple mode, and weighted mode estimators. This was done to verify the concordance of causal results derived from distinct statistical approaches. A series of sensitivity analyses were executed to verify the stability of our causal deductions and uncover possible breaches of the fundamental MR hypotheses. We performed MR-Egger regression to screen for directional horizontal pleiotropy; in this context, an intercept that deviates from zero indicates potential bias. Furthermore, we applied Cochran's Q test to examine heterogeneity across the instruments, where a p-value <0.05 was interpreted as indicating a lack of uniformity in the effects of the instruments. Moreover, leave-one-out sensitivity checks were performed to pinpoint any individual SNP that could be exerting an excessive influence on the aggregate causal estimation. 37 Collectively, these verification steps substantiated that the derived causal estimates were not artifacts of pleiotropy or heterogeneity, thereby strengthening the reliability of the MR methodology employed.
Results
Treg-specific genetic regulation of RA
Among the nine Treg-specific cis-eQTLs evaluated, two loci reached nominal statistical significance (p < 0.05). The DDX60L locus conferred an elevated risk of RA (odds ratio (OR) = 2.10, 95% confidence interval (CI): 1.10–3.98, p = 0.024), indicating that each genetically predicted 1-SD increase in DDX60L expression in Tregs raises disease odds by approximately 110%. Conversely, the OAS1 locus exerted a protective effect (OR = 0.65, 95% CI 0.45–0.94, p = 0.021), implying that higher Treg-specific OAS1 expression lowers the risk by 35%. The remaining seven genes (CEP135, HLA-A, IFITM3, JAKMIP2, SLC25A3, TXNRD1, and VPS9D1) showed non-significant estimates (p > 0.05), with ORs centered around the null (Figure 1). To test whether the causal links hold under varying analytical settings and probe possible breaches of MR prerequisites, we performed a battery of sensitivity tests. The scatter plot shows MR estimates for OAS1, with the x-axis representing the effects of SNPs on OAS1 expression and the y-axis showing their effects on RA (Figure 2(a)). Cochran’s Q tests indicated no significant heterogeneity among the IVs (p > 0.05). Leave-one-out analyses further confirmed that no single SNP exerted a disproportionate influence on the overall causal estimate, thereby strengthening confidence in the robustness of the MR findings (Figure 2(b)). Apart from that for OAS1, heterogeneity and pleiotropy analyses could not be conducted due to an insufficient number of SNPs. These findings provide preliminary evidence that genetically regulated expressions of DDX60L and OAS1 within Tregs may promote and inhibit RA susceptibility, respectively.
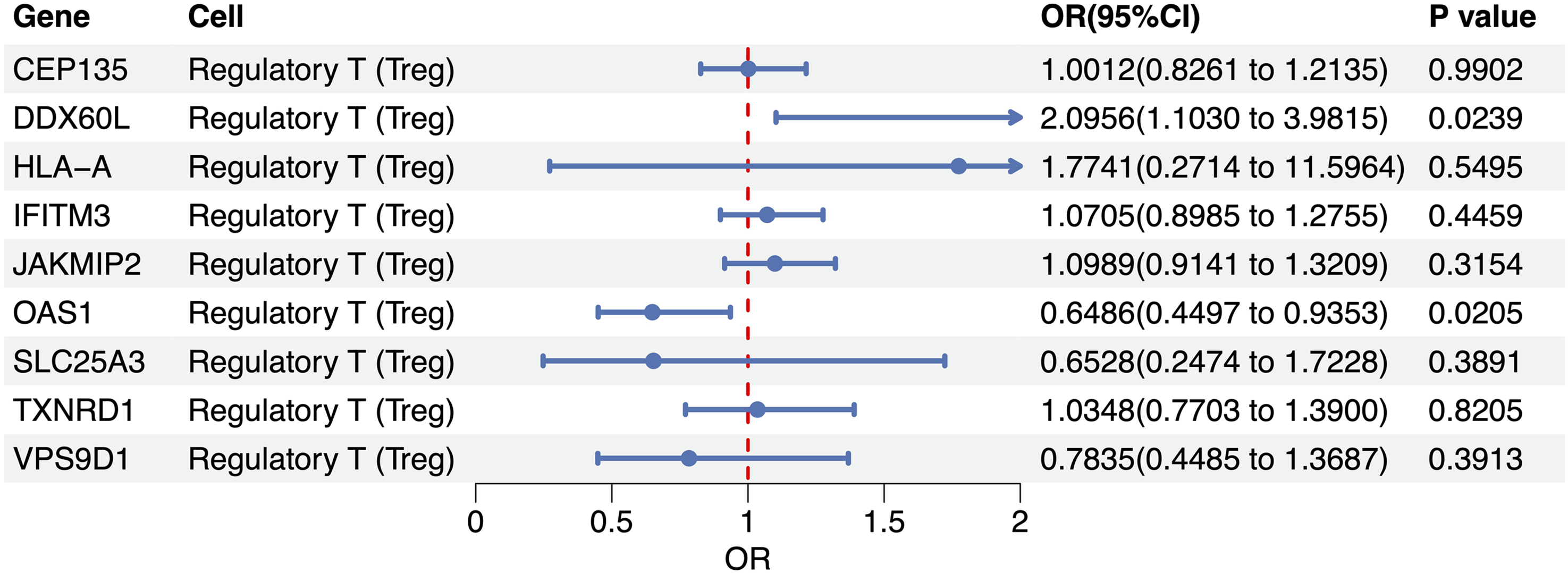
Treg-specific cis-eQTL for rheumatoid arthritis.

Robustness checks for the MR findings. (a) IVW-derived causal estimates are visualized in scatter plots for selected Treg-restricted cis-eQTLs; (b) leave-one-out validation confirms that no single variant drives the observed causal links.
Treg-specific genetic regulation of GA
Among the eight Treg-specific cis-eQTLs examined, two loci showed statistically significant associations with GA. The OAS1 expression in Tregs exhibited a robust protective effect (OR = 0.897, 95% CI: 0.860–0.936, p < 0.001), indicating that each genetically predicted 1-SD increase in the OAS1 abundance reduced the knee osteoarthritis risk by approximately 10%. In contrast, JAKMIP2 conferred a modest but significant increase in disease susceptibility (OR = 1.019, 95% CI: 1.001–1.038, p = 0.044), corresponding to 1.9% higher odds per 1-SD increment in OAS1 abundance. The remaining six loci (HLA-A, IFITM3, SLC25A3, SULF2, TXNRD1, and VPS9D1) yielded directionally heterogeneous and non-significant estimates (p ≥ 0.08), with confidence intervals spanning the null (Figure 3). To test whether the causal links hold under varying analytical settings and probe possible breaches of MR prerequisites, we performed a battery of sensitivity tests. The scatter plot shows MR estimates for OAS1, with the x-axis representing the effects of SNPs on OAS1 expression and the y-axis showing their effects on GA (Figure 4(a)). Cochran’s Q tests indicated no significant heterogeneity among the IVs (p > 0.05). Leave-one-out analyses further confirmed that no single SNP exerted a disproportionate influence on the overall causal estimate, thereby strengthening confidence in the robustness of the MR analysis (Figure 4(b)). Except for OAS1, heterogeneity and pleiotropy analyses could not be conducted due to an insufficient number of SNPs. These findings provide the first genetically anchored evidence that Treg-specific modulation of OAS1 and JAKMIP2 may confer protection against and predisposition to GA, respectively.
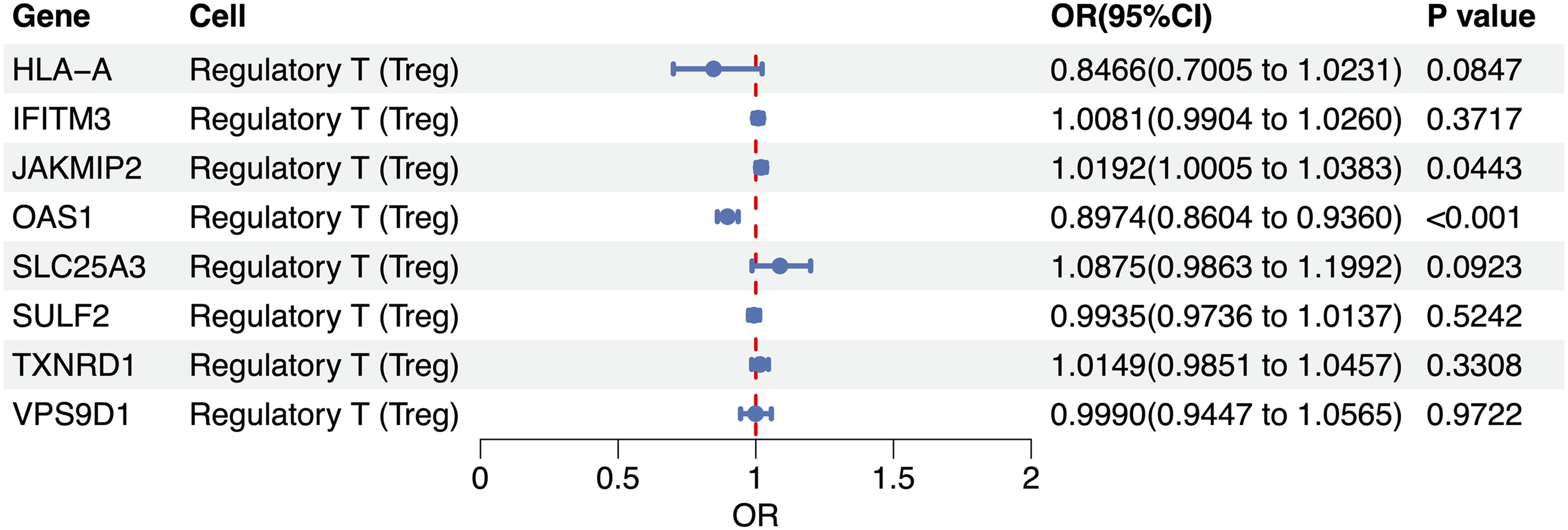
Treg-specific cis-QTL for gonarthrosis.

Robustness checks for the MR findings. (a) IVW-derived causal estimates are visualized in scatter plots for selected Treg-restricted cis-eQTLs; (b) leave-one-out validation confirms that no single variant drives the observed causal links.
Treg-specific genetic regulation of GOA
Among the ten Treg-specific cis-eQTLs assessed, none surpassed the nominal significance threshold (p < 0.05) for GOA. The tightest confidence interval surrounded OAS1, the genetically predicted expression of which suggested a modest but non-significant protective trend (OR = 0.938, 95% CI: 0.872–1.009, p = 0.086). Conversely, CEP135 and JAKMIP2 yielded marginally positive point estimates (OR = 1.022 and 1.016, respectively), whereas the remaining loci—DDX60L, HLA-A, IFITM3, SLC25A3, SULF2, TXNRD1, and VPS9D1—clustered closely around the null (OR 0.82–1.07, all p > 0.26, Figure 5). To test whether the causal links hold under varying analytical settings and probe possible breaches of MR prerequisites, we performed a battery of sensitivity tests. The scatter plot shows MR estimates for OAS1, with the x-axis representing the effects of SNPs on the OAS1 expression and the y-axis showing their effects on GOA (Figure 6(a)). Cochran’s Q tests indicated no significant heterogeneity among the IVs (p > 0.05). Leave-one-out analyses further confirmed that no single SNP exerted a disproportionate influence on the overall causal estimate, thereby strengthening confidence in the robustness of the MR findings (Figure 6(b)). Except for OAS1, heterogeneity and pleiotropy analyses could not be conducted due to an insufficient number of SNPs.
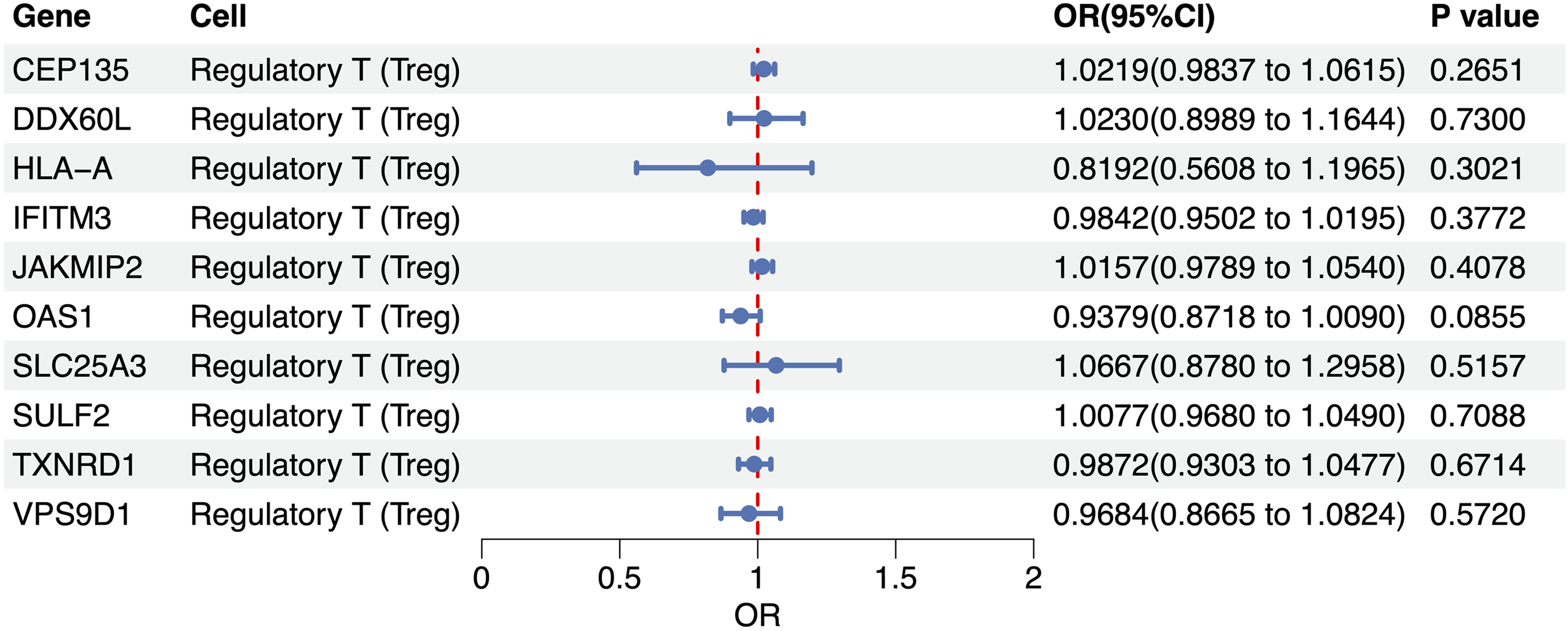
Treg-specific cis-eQTL for gouty arthritis.
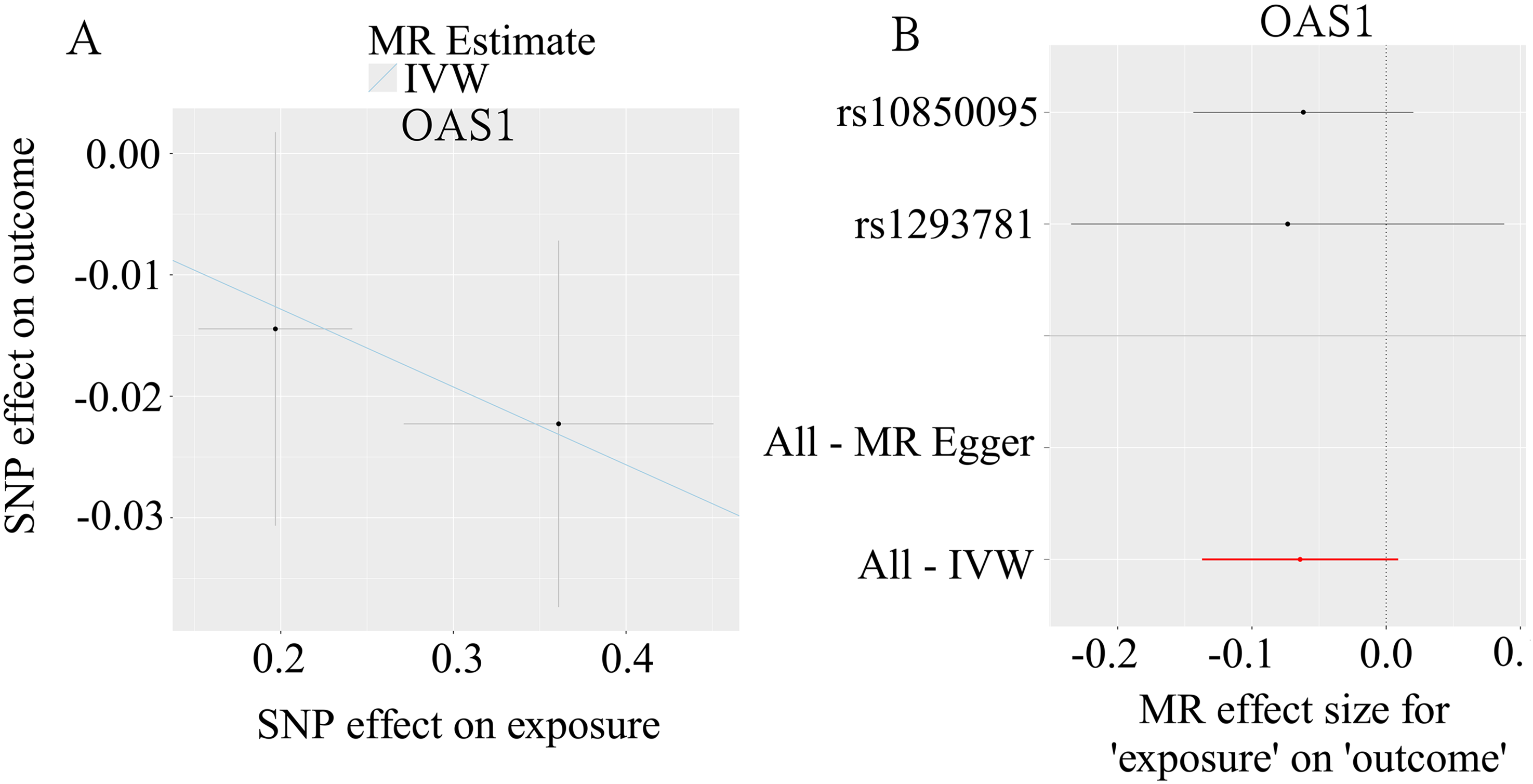
Robustness checks for the MR findings. (a) IVW-derived causal estimates are visualized in scatter plots for selected Treg-restricted cis-eQTLs; (b) leave-one-out validation confirms that no single variant drives the observed causal links.
This study integrated sc–cis-eQTLs with two-sample MR analysis to systematically assess the causal impact of Treg-specific gene expression on three types of arthritides. In RA, DDX60L increased risk whereas OAS1 was protective; in GA, OAS1 remained protective and JAKMIP2 modestly raised the risk; GOA did not demonstrate any significant associations. Collectively, OAS1 emerges as a cross-disease guardian, whereas DDX60L and JAKMIP2 may require disease-tailored modulation, providing genetic guidance for developing universal Treg therapies characterized by high OAS1 and low DDX60L/JAKMIP2 activity (Figure 7).
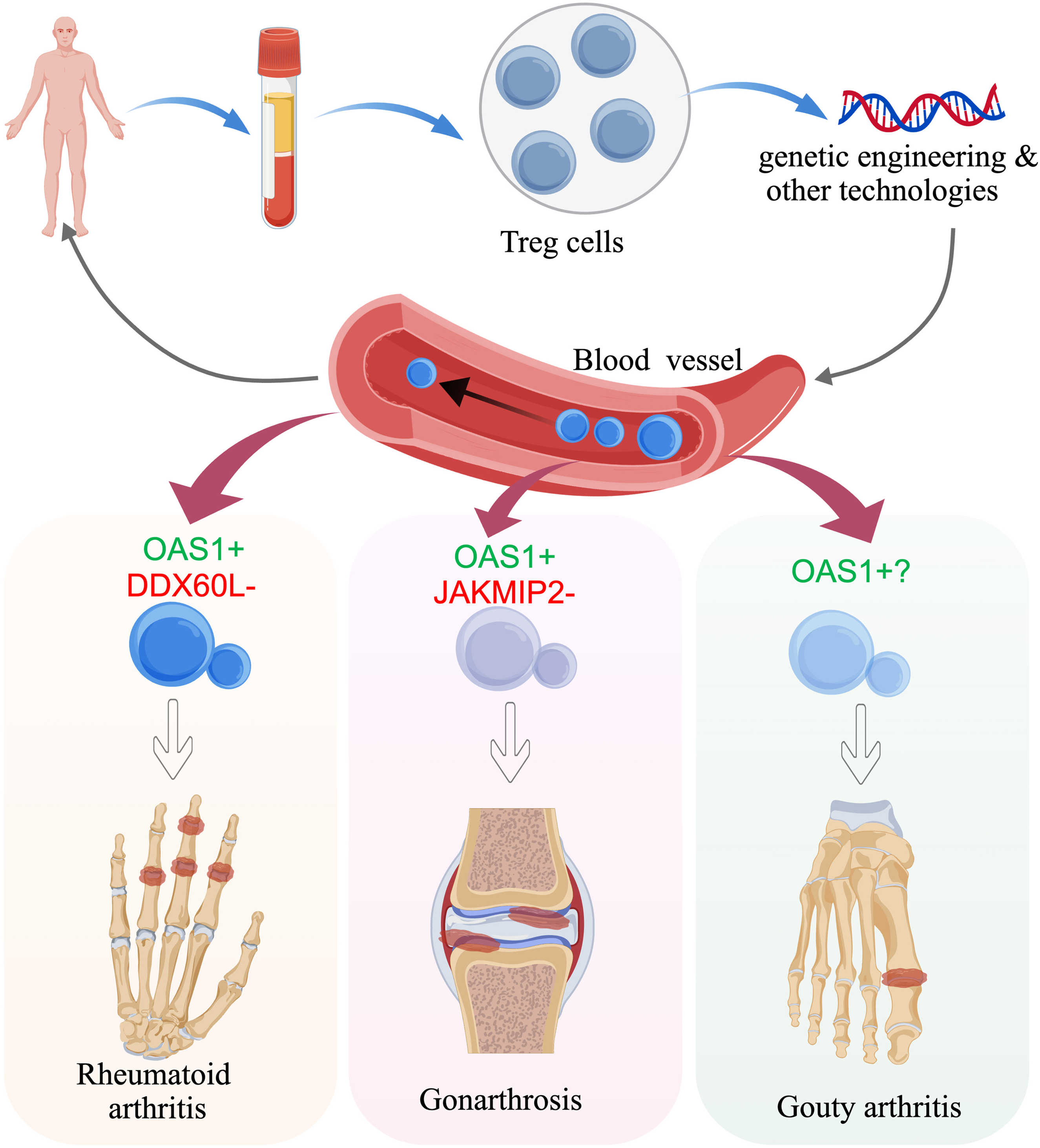
Single-cell cis-eQTL–MR integrative profiling identifies high-priority genetic candidates for RA, GA and GOA therapy. The graphic color-codes protective signals (green for OAS1) and risk signals (red for DDX60L/JAKMIP2) and illustrates CRISPR or CAR-Treg approaches to “turn up green, silence red.” When engineering a universal Treg therapy for all three diseases, prioritize a subset with high OAS1 and low DDX60L/JAKMIP2 expression; OAS1 is the common protector, whereas DDX60L and JAKMIP2 require disease-specific modulation.
Discussion
In this study, we conducted a comprehensive integration of single-cell Treg cis-eQTLs and two-sample MR to elucidate the causal impact of Treg-specific gene expression on the pathogenesis of three distinct forms of arthritis: (a) RA; (b) GA; and (c) GOA. By leveraging high-resolution genetic instruments, we successfully identified key molecular targets within the Treg transcriptome that exert significant causal influences on disease susceptibility. Our findings reveal a complex landscape where the OAS1 gene acts as a universal protector across RA and GA, whereas DDX60L and JAKMIP2 serve as context-specific risk factors for RA and GA, respectively. Notably, no significant Treg-specific causal signals were detected for GOA, suggesting distinct immunological drivers for this crystal-induced pathology.
The identification of OAS1 as a robust protective factor in both RA and GA represents a critical advancement in our understanding of Treg-mediated immune tolerance. OAS1 is a well-characterized interferon-stimulated gene classically involved in the antiviral response via the activation of ribonuclease L (RNase L). 38
In addition to this, OAS1 plays non-canonical roles, including promoting interferon regulatory factor 1 messenger (IRF1) messenger ribonucleic acid (mRNA) translation to enhance antibacterial immunity, 39 protecting interferon beta (IFNβ) mRNA from degradation by binding adenylate–uridylate (AU)–rich elements, 40 and suppressing African swine fever virus by recruiting tripartite motif containing 21 (TRIM21) to degrade the viral capsid protein P72. 41 Although its role in viral defense is established, its function in modulating autoimmune or degenerative inflammation through Tregs is less explored. Our MR analysis indicates that genetically predicted higher expression of OAS1 in Tregs significantly reduces the risk of RA and GA. In RA, the presence of an IFN-I signature is well documented, particularly in early, treatment-naïve patients, where IFN-α correlates strongly with the interferon gene signature in whole blood. 42 Although direct evidence linking OAS1 upregulation in Tregs to functional preservation is not explicitly detailed in the provided materials, it is established that Treg dysfunction and reduced suppressive capacity are central to RA immunopathology.43,44 In RA, Tregs exhibit phenotypic instability, including increased frequencies of Th1- and Th17-like Tregs, which are associated with disease activity.43,45 Given that IFN-I signaling can modulate T cell responses and that OAS1 is a canonical IFN-stimulated gene, it is crucial to explicitly distinguish the causal genetic evidence established in our MR analysis from the mechanistic hypotheses that follow. Although our findings robustly demonstrate that genetically predicted higher OAS1 expression in Tregs reduces disease risk, the biological pathways mediating this protective effect remain to be determined. Therefore, we speculate that OAS1 induction in Tregs may serve to limit excessive inflammatory signaling or promote Treg survival under conditions of inflammatory stress. This could theoretically help maintain a functional Treg pool capable of suppressing pathogenic Th1 and Th17 responses, which are key drivers of synovial inflammation and tissue damage in RA.46,47 This proposed mechanistic link, although biologically plausible, is strictly inferential and necessitates direct functional validation in future studies. Similarly, in GA, a disease driven by low-grade inflammation and mechanical stress, OAS1-enhanced Tregs may more effectively modulate macrophage polarization and inhibit the production of matrix metalloproteinases, thus protecting cartilage integrity. 48 The cross-disease efficacy of OAS1 positions it as a prime candidate for “universal” Treg engineering strategies, offering a potential therapeutic avenue that transcends specific pathological classifications.
Conversely, our results highlight DDX60L, as a specific risk factor for RA. Genetically predicted upregulation of DDX60L in Tregs was associated with a substantial increase in the RA risk. DDX60L is a DexD/H-box RNA helicase belonging to a family of proteins known to participate in antiviral innate immunity. 49 However, direct functional evidence for DDX60L in this context remains limited; most available studies have focused on its oncogenic properties in cancer. In particular, high DDX60L expression is linked to poor prognosis in hepatocellular carcinoma and pancreatic ductal adenocarcinoma, where it promotes tumor progression, metastasis, and modulates the tumor microenvironment.50,51 Currently, no direct evidence links DDX60L to the regulation or function of Tregs. Its known roles are primarily in viral sensing, RNA metabolism, and tumor-intrinsic signaling pathways rather than adaptive immune cell modulation.50,51 Therapeutic strategies aimed at silencing DDX60L or modulating its downstream effectors specifically within Tregs may help refine the management of RA, offering a targeted approach to restore immune regulation without broad immunosuppression.
In the context of GA, JAKMIP2 emerged as a modest but significant risk factor. JAKMIP2 is a scaffold protein primarily involved in the regulation of microtubule dynamics and intracellular signaling. 52 JAKMIP2 also modulates JAK/STAT signaling by interacting with JAK kinases, potentially affecting downstream inflammatory and proliferative pathways. 53 Although its role in immune and neuronal cells has been more extensively studied, emerging evidence suggests involvement in musculoskeletal biology. Currently, there is no direct evidence linking JAKMIP2 to osteoarthritis pathogenesis. However, given its interaction with the JAK2/STAT3 pathway—a key regulator of cartilage degradation, synovial inflammation, and subchondral bone remodeling in osteoarthritis (OA);54,55 JAKMIP2 may indirectly influence OA progression via the modulation of JAK/STAT activity. 54 Further research is needed to determine whether JAKMIP2 expression or function is altered in OA tissues and whether it contributes to dysregulated JAK signaling in chondrocytes or synovial cells.
A significant observation in this study is the lack of significant causal signals for GOA. Among the evaluated Treg-specific cis-eQTLs, none reached nominal significance for GOA. The strongest signal, OAS1, showed only a non-significant protective trend, potentially reflecting the distinct pathophysiology of gout, which is primarily driven by NLR family pyrin domain containing 3 (NLRP3) inflammasome activation in response to monosodium urate crystals,56,57 involving neutrophils and macrophages rather than adaptive T cell dysregulation. Although Tregs are known to play a role in resolving gouty inflammation, 58 our genetic analysis suggests that inherited, basal differences in Treg-specific gene expression (as captured by cis-eQTLs) are not a primary determinant of gout susceptibility.
The methodological integration of single-cell eQTLs with MR constitutes a major strength of this study. Bulk eQTL analyses often obscure cell type–specific regulatory mechanisms due to tissue averaging. By utilizing single-cell resolution data, we successfully isolated the genetic regulation specific to Tregs, confirming the hypothesis that cis-eQTLs in Tregs are distinct from those in other immune cell types. This specificity is crucial for identifying precise therapeutic targets. If we had relied on bulk data, the nuanced effects of DDX60L or JAKMIP2 might have been masked. The MR framework further strengthened our inference by minimizing confounding and reverse causation, providing robust evidence for the causal role of these genes in arthritis pathogenesis. The rigorous sensitivity analyses, including heterogeneity tests and leave-one-out validation, further confirmed the reliability of our estimates, particularly for the OAS1 findings. These findings have profound implications for the future of Treg-based immunotherapy. Current clinical efforts are exploring the infusion of ex vivo–expanded Tregs or the use of CAR-Tregs to treat autoimmune diseases. However, a major challenge is ensuring that these cells maintain potent suppressive function and stability in the inflammatory joint environment. Our genetic blueprint suggests that next-generation Treg therapies should be engineered to possess a “gain-of-function” profile regarding protective genes such as OAS1 and a “loss-of-function” profile regarding risk genes such as DDX60L and JAKMIP2. For instance, utilizing clustered regularly interspaced short palindromic repeats (CRISPR) activation to upregulate OAS1 or CRISPR interference to silence DDX60L could create “armored” Treg products with enhanced resilience against inflammatory triggers.
Furthermore, to overcome the scalability and in vivo stability challenges inherent to live-cell therapies, this genetic blueprint can be directly translated into cell-free modalities. Engineering allogeneic Treg-derived vesicles—such as HLA class I–knockdown artificial vesicles that retain key immunosuppressive proteins and effectively suppress pathogenic cytokine secretion—offers a highly promising, scalable, and off-the-shelf immunotherapeutic alternative. 59 Applying our high-OAS1/low-DDX60L/JAKMIP2 signature to engineer such optimized Treg-derived vesicles could provide a next-generation, cell-free precision therapy for arthritis. Additionally, for a live “universal” Treg therapy designed to treat both RA and GA, the ideal cellular product would be one with high OAS1 expression and low DDX60L/JAKMIP2 activity, as proposed in our schematic model (Figure 7). This precision engineering approach addresses the heterogeneity of arthritis and moves beyond the “one-size-fits-all” paradigm.
Certain study limitations must be acknowledged. First, the MR analysis relies on the assumptions of no horizontal pleiotropy; although we performed sensitivity analyses, residual pleiotropic effects cannot be entirely ruled out. Second, the sc-eQTL data, although high-resolution, may not capture the full spectrum of Treg heterogeneity, such as differences between resting and activated Tregs, or tissue-resident versus circulating Tregs. Third, our study population was primarily of European ethnicity; therefore, the generalizability of these genetic associations to other ethnic groups requires further validation. Additionally, the functional mechanisms by which OAS1, DDX60L, and JAKMIP2 modulate Treg behavior were inferred genetically and require direct validation through in vitro and in vivo experiments.
Taken together, this study bridges the gap between immunogenetics and bioengineering by delineating the causal architecture of Treg-specific gene regulation in arthritis. We identified OAS1 as a shared protective agent, whereas DDX60L and JAKMIP2 act as disease-specific risk amplifiers in RA and GA, respectively. These insights provide a rational genetic basis for the design of enhanced Treg therapies, highlighting the potential of genetic modulation to optimize therapeutic outcomes. Future research should focus on functional validation of these targets and the development of gene-edited Treg products, paving the way for personalized, mechanism-driven treatments for debilitating arthritic conditions.
Conclusions
We identified OAS1, DDX60L, and JAKMIP2 as causal Treg-specific modulators that delineate the genetic architecture of arthritis susceptibility. As CAR-Treg therapies advance toward early-phase clinical trials, these genes serve as precise targets for CRISPR-mediated “armoring” to overcome the bottleneck of cell-intrinsic functional instability. To translate this genetic blueprint into clinical reality, it is imperative to conduct targeted in vitro and in vivo validations confirming the suppressive durability and tissue-homing precision of these edited Tregs within the RA and GA joint microenvironments.
Footnotes
Acknowledgments
We thank the FinnGen consortium and the scQTLbase database for providing open-access GWAS summary statistics and single-cell eQTL data. We are grateful to all investigators and participants who contributed to these datasets.
Author contributions
Tianqi Ren, Yiwei Shen, and Ji Li. mainly contributed to the primary design and execution of formal analyses and authored the initial draft of the manuscript. Zhe Zhang, Zelin Liu, and Lei Bao undertook data curation and validated the dataset. Tianqi Ren and Xue Li contributed to manuscript revision and scrutinized the results. Yiwei Shen and Ji Li supervised the entire project. All the authors have thoroughly reviewed, critically discussed, and reached a consensus on the final version of the manuscript for publication.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.