
Abstract
BK polyomavirus (BKPyV) is highly prevalent and clinically relevant in immunocompromised populations, underscoring the need for reliable serological tools to support epidemiological monitoring and clinical follow-up. This study aimed to generate a recombinant VP1-based enzyme-linked immunosorbent assay (ELISA) using a baculovirus–Sf9 insect cell system and to explore its application in a human serum cohort. Recombinant VP1 was produced by baculovirus-mediated expression, purified by nickel–nitrilotriacetic acid affinity chromatography, and used to establish an indirect IgG ELISA. A plasmid-based transient expression approach in Sf9 cells was evaluated only as a feasibility experiment and did not yield sufficient VP1 for assay development. Analytical performance was assessed using a polymerase chain reaction (PCR)-defined serum panel (n = 30), and the assay was subsequently applied to 149 sera with unknown BKPyV status. Baculovirus-mediated expression generated high-purity VP1 with an average yield of approximately 1.55 mg/mL from a 10 mL culture. PCR-confirmed positive sera showed markedly higher optical density values than PCR-negative sera, with very good analytical discrimination in this small validation set (area under the curve = 0.996). In the cohort analysis, overall seroreactivity was 89.7%, antibody reactivity increased with age, and higher ELISA signals were observed in renal transplant and hemodialysis patients than in healthy individuals. Detectable reactivity in young children may reflect early exposure or passive maternal antibodies. These findings indicate that insect cell–derived BKPyV VP1 is a suitable antigen for serological assays and a practical platform for preliminary seroepidemiological applications, which require confirmation in larger independent validation cohorts.
Introduction
BK polyomavirus (BKPyV), first characterized in 1971 from a kidney transplant patient, is an ordinary member of the Polyomaviridae family with a circular double-stranded DNA genome and the potential for lifelong latent infection of renal epithelial cells of immunocompetent hosts (Helle et al., 2017; Zhou et al., 2023; Nourie et al., 2024). Although primary infection is typically asymptomatic, viral reactivation under immunosuppressive conditions, as seen in kidney and hematopoietic stem cell transplant recipients, can result in BKPyV-associated nephropathy (BKVAN) and hemorrhagic cystitis, both of which are major causes of graft dysfunction and clinical morbidity (Bennett et al., 2012; Moens et al., 2014; Nagshabandi, 2020; Furmaga et al., 2021; Kiasari et al., 2024; Nourie et al., 2024). Furthermore, there is cumulative evidence to indicate that BKPyV is an oncogenic cofactor for various cancers, such as urinary tract, prostate, and head and neck cancers (Tognon et al., 2003; Burger-Calderon and Webster-Cyriaque, 2015; Levican et al., 2018; Kotton et al., 2024; AlMalaki et al., 2025; Shishkova et al., 2025).
Given the high global seroprevalence of BKPyV and the lack of effective antiviral treatments or licensed vaccines, precise and sensitive diagnostic techniques are needed for monitoring viral reactivation and informing clinical management (Höcker et al., 2019; Zhou et al., 2020; Kausar et al., 2025). Although nucleic acid-based techniques have made great strides in BKPyV detection and genotyping, serology remains necessary for understanding long-term immunity and infection patterns (Govind et al., 2019; Ahlenstiel-Grunow and Pape, 2020; Furmaga et al., 2021; Bahrulolum et al., 2023). The major capsid protein VP1, forming the outer surface of the virion and containing key conformational epitopes, is the primary antigenic target for serodiagnostic assay development (Salunke et al., 1989; Gillock et al., 1997; Chen et al., 1998).
Conventional recombinant VP1 production using Escherichia coli or yeast systems often results in misfolded protein lacking native conformational epitopes (Srivastava et al., 2023; Sookhoo et al., 2024). In contrast, baculovirus–insect cell expression systems support high-level expression and may facilitate correct protein folding and assembly; however, the extent of higher-order assembly depends on the production and purification strategy and requires dedicated structural characterization (Jarvis, 2009; Van Oers, 2011; Contreras-Gómez et al., 2014). Although baculovirus production offers clear advantages, it also involves multi-step virus generation and amplification. Plasmid-based transient expression in insect cells is simpler but generally produces low yields and may not preserve antigenic integrity. Despite the extensive use of both strategies, direct experimental comparisons for BKPyV VP1 production remain limited (Hitchman et al., 2009; Van Oers, 2011).
To address these challenges and facilitate the development of reliable serological assays, this study focused primarily on producing high-quality recombinant BKPyV VP1 using a baculovirus–insect cell system and applying it to a human cohort to assess BKPyV seroprevalence across age and clinical groups. This work provides a practical recombinant antigen platform to support BKPyV serodiagnosis and epidemiological monitoring. However, higher-order assembly of VP1 was not directly assessed in this study, and antigenic authenticity is therefore inferred from serological performance rather than confirmed structurally.
Materials and Methods
Human serum samples collection
This study utilized two distinct groups of human serum samples. Group A consisted of 30 sera used for analytical evaluation of the ELISA, including 15 samples obtained from individuals with BKPyV DNA confirmed by polymerase chain reaction (PCR) and collected during the convalescent phase (2–4 weeks after symptom onset), and 15 PCR-negative control sera obtained from asymptomatic individuals with no reported respiratory or urological illness within the preceding year. These samples were used exclusively for initial assay optimization and performance assessment. Group B comprised 149 de-identified human serum samples with unknown BKPyV exposure status, selected from an available epidemiological serum repository spanning an age range of 1–60 years (mean age: 28 years; median age: 32 years). No stratified or population-based sampling framework was applied, and samples were included without prior BKPyV screening. This cohort was used to explore serological reactivity patterns across age and clinical subgroups rather than to derive population-level prevalence estimates.
All serum samples were collected between September 2024 and April 2025, anonymized in accordance with institutional ethical guidelines, and stored at −70°C until analysis. Written informed consent was obtained from all participants or their legal guardians, and the study protocol was approved by the relevant institutional review board.
PCR amplification of the BKPyV VP1 region
To amplify the BKPyV VP1 gene, conserved nucleotide regions were identified through multiple sequence alignment of publicly available BKPyV strains using BioEdit software (Ibis Biosciences, USA). Based on these alignments, specific primer pairs were designed with 5′ overhangs to enable directional cloning into the expression vector (Table 1). Primer specificity was verified in silico using the NCBI BLAST tool. Viral DNA was extracted from PCR-confirmed BKPyV-positive clinical specimens and used as the template for amplification of the VP1 coding region using a high-fidelity PCR protocol to minimize sequence errors. The resulting amplicons were purified and analyzed by 1% agarose gel electrophoresis to confirm the expected product size and integrity prior to downstream cloning and expression procedures.
Primers Used for Amplification of the BKPyV VP1 Gene

Generation of recombinant expression vector
The amplified BKPyV VP1 DNA fragment were cloned into the pIEx/Bac-3 3 C/LIC expression vector (Novagen, USA), which is designed for high-level protein expression in insect cell lines under the control of the AcNPV hr5 enhancer and ie1 promoter elements. Ligation-independent cloning (LIC) was performed using T4 DNA polymerase–mediated end processing of both the VP1 insert and the linearized vector to generate compatible overhangs for directional cloning. Following annealing, the recombinant constructs were transformed into competent Escherichia coli NovaBlue GigaSingles™ cells. Positive transformants were screened by colony PCR to verify the presence of the VP1 insert, and recombinant plasmids were subsequently purified using the Mobius™ 200 Plasmid Kit (Novagen). Successful insertion of the VP1 coding sequence was confirmed by agarose gel electrophoresis (1%) prior to downstream expression experiments.
Recombinant VP1 protein expression in Sf9 insect cells
Recombinant BKPyV VP1 expression was primarily achieved using a baculovirus–insect cell expression system, which served as the main platform for antigen production. In parallel, a plasmid-based transient transfection approach was evaluated solely as an exploratory assessment of expression feasibility and was not intended to represent a fully comparative production system. For transient expression, Sf9 cells were seeded at a density of 1 × 106 cells/mL in serum-free BacVector™ medium and transfected with 20 µg of recombinant pIEx/Bac-3–VP1 plasmid using 100 µL of Insect GeneJuice™ transfection reagent (Novagen), according to the manufacturer’s instructions. Transfected cells were incubated at 28°C with gentle agitation (150 rpm) for 48 h prior to analysis. For baculovirus-mediated expression, Sf9 cells were cotransfected with 500 ng of the recombinant VP1 expression plasmid and 100 ng of BacMagic™ baculoviral DNA using 5 µL of Insect GeneJuice™ in 1 mL of culture medium. Recombinant baculovirus particles were harvested five days post-transfection and subsequently amplified. For large-scale protein production, Sf9 cells (1 × 108 cells) were infected with the amplified recombinant baculovirus at a multiplicity of infection (MOI) of 10 in 50 mL of fresh medium and incubated at 28°C for 72 h under continuous shaking conditions.
Protein extraction and purification
Sf9 insect cells infected with recombinant baculovirus were harvested and lysed using 20 mL of CytoBuster™ protein extraction reagent (Novagen), followed by incubation for 5 min at room temperature. Cell debris was removed by centrifugation at 5,000 × g for 10 min, and the resulting clarified lysate was subjected to affinity purification. His-tagged VP1 protein were purified from the clarified lysate using Ni–NTA His•Bind® affinity resin according to the manufacturer’s protocol. After extensive washing to remove nonspecifically bound proteins, recombinant VP1 was eluted using an imidazole-containing buffer. Protein concentration was determined spectrophotometrically by UV absorbance measurements, and purified fractions were used directly for downstream immunological assays without further structural fractionation.
Immunofluorescence microscopy
For immunofluorescence analysis, Sf9 cells expressing recombinant BKPyV VP1 were fixed on polytetrafluoroethylene (PTFE)-coated slides using ice-cold acetone (−20°C). Fixed cells were blocked with a serum-containing blocking buffer and incubated with a monoclonal antibody specific for VP1 (1:100 dilution). Following washing steps, cells were incubated with fluorescein isothiocyanate–conjugated secondary antibodies. Fluorescent signals were visualized using a fluorescence microscope at 400 × magnification to confirm intracellular expression and antigen-specific antibody recognition.
SDS-PAGE and immunoblot analysis
Purified recombinant VP1 protein samples were separated on 4–12% bis-Tris sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under denaturing conditions. For immunoblot analysis, resolved proteins were transferred onto polyvinylidene difluoride membranes and probed with anti-VP1 primary antibodies (1:1500 dilution), followed by horseradish peroxidase (HRP) or alkaline phosphatase-conjugated secondary antibodies. Immunoreactive bands were visualized using 3,3′-diaminobenzidine (DAB) or 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) chromogenic substrates.
Dot blot analysis was additionally performed as a qualitative and semi-quantitative approach to assess VP1 immunoreactivity. These immunochemical assays confirmed expression and basic immunoreactivity of recombinant VP1. They do not provide information regarding pentamerization or virus-like particle (VLP) formation, which were beyond the scope of the present work.
ELISA development for serological detection
High-binding 96-well microtiter plates (Immulon 2, Dynex Technologies) were coated overnight at 4°C with purified recombinant BKPyV VP1 antigen (0.2 µg per well) diluted in phosphate-buffered saline (PBS; pH 7.2). Plates were subsequently blocked with 5% (w/v) skim milk in PBS containing 0.05% Tween-20 (PBS-T) for 1 h at room temperature to reduce nonspecific binding. Serum samples were diluted 1:40 in blocking buffer and incubated in duplicate wells at 37°C for 1 h. After washing with PBS-T, HRP–conjugated anti-human IgG antibody (1:4,000 dilution; Sigma, UK) was added and incubated for 30 min at room temperature. Colorimetric detection was performed using ABTS substrate, and absorbance was measured at 405 nm using a microplate reader. The ELISA cutoff value was initially defined as the mean optical density (OD) of PCR-confirmed BKPyV-negative control sera plus three standard deviations (mean + 3SD). To further evaluate assay discrimination and performance, receiver operating characteristic (ROC) curve analysis were applied using the PCR-defined serum panel. Samples yielding OD values within ± 10% of the calculated cutoff were considered borderline and were retested in duplicate to minimize misclassification.
Statistical analysis
Statistical analyses were performed using SPSS v26. Descriptive data are reported as mean ± SD and/or median (range), as appropriate. Normality of ELISA OD405 values were evaluated using the Shapiro–Wilk test; therefore, non-parametric tests were applied for group comparisons. The Mann–Whitney U test was used for two-group OD comparisons, and Kruskal–Wallis test was used for comparisons across multiple age groups. ROC curve analysis was performed on the PCR-defined panel (n = 30) to evaluate analytical discrimination, reporting the area under the curve (AUC) (95% CI) and determining the optimal cutoff using the Youden index. For cohort seroreactivity classification, the cutoff was defined as mean OD of PCR-negative sera + 3SD. All tests were two-tailed, and p < 0.05 was considered statistically significant.
Results
The sequence of experimental steps leading from VP1 production to analytical evaluation and application to the human serum cohort is shown schematically in Figure 1.
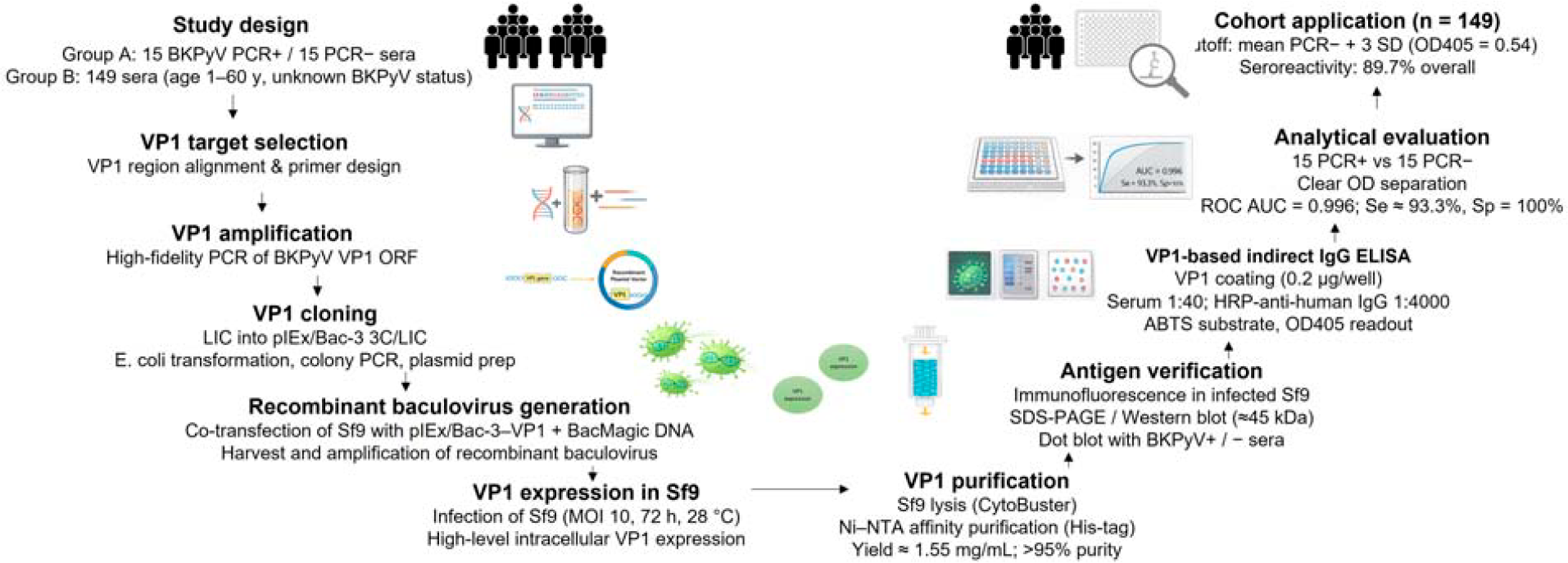
Schematic overview of study design, VP1 production workflow, assay development, and application. Schematic summary of BKPyV VP1 production and ELISA development. The study included a PCR-defined validation panel (15 BKPyV-positive, 15 BKPyV-negative sera) and a cohort of 149 sera with unknown status. The VP1 ORF was amplified, cloned into pIEx/Bac-3 3C/LIC, and used to generate recombinant baculovirus for infection of Sf9 cells. Following intracellular expression, VP1 was purified by Ni–NTA affinity chromatography (yield 1.55 mg/mL; >95% purity). Antigen expression and immunoreactivity were confirmed by immunofluorescence, SDS-PAGE, Western blotting, and dot blotting. An indirect VP1-based IgG ELISA was established and analytically evaluated (ROC AUC 0.996; Se 93.3%; Sp 100%) before application to the cohort, yielding an overall seroreactivity of 89.7% using a cutoff of mean PCR ± 3SD (OD405 = 0.54). AUC, area under the curve; BKPyV, BK polyomavirus; LIC, L igation-independent cloning; Ni–NTA, nickel–nitrilotriacetic acid; PCR, polymerase chain reaction; ROC, receiver operating characteristic.
The BKPyV major capsid protein VP1 was successfully expressed in Sf9 insect cells following infection with a recombinant baculovirus encoding the VP1 gene. Indirect immunofluorescence analysis demonstrated strong intracellular fluorescence signals in baculovirus-infected Sf9 cells at 72 h postinfection, indicating efficient expression of VP1 (Fig. 2). In contrast, mock-infected control cells showed no detectable fluorescence, confirming the specificity of VP1 expression and antibody recognition. Recombinant VP1 expression was further confirmed by immunochemical analyses. SDS-PAGE analysis of cell lysates from baculovirus-infected Sf9 cells revealed a distinct protein band at approximately 45 kDa, consistent with the expected molecular weight of BKPyV VP1. Western blot analysis using a VP1-specific monoclonal antibody confirmed a corresponding immunoreactive band, whereas no signal was detected in uninfected control cell lysates. Dot blot assays demonstrated strong reactivity between recombinant VP1 and BKPyV-positive human sera, while negative control sera showed no detectable binding, supporting the presence of VP1-specific linear and potentially some conformational epitopes, although higher-order structural assembly was not assessed (Fig. 3). In parallel, a plasmid-based transient transfection approach was evaluated as an exploratory expression strategy in Sf9 cells. Under the tested conditions, VP1 expression levels were minimal, with no detectable signal observed by SDS-PAGE, Western blotting, or dot blot analysis. Due to insufficient antigen production, this approach was not pursued further for downstream immunoassay development (Table 2).
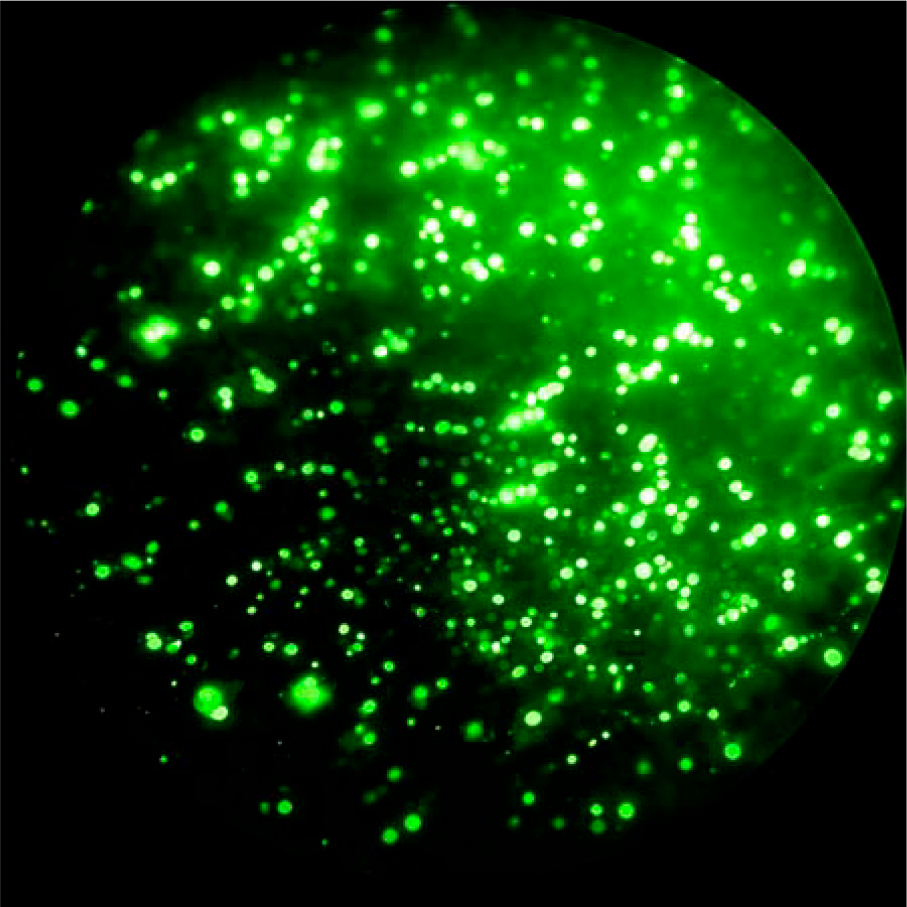
Immunofluorescence detection of recombinant BK polyomavirus VP1 expression in Sf9 insect cells following baculovirus-mediated infection. Cells were fixed at 72 h postinfection (p.i.) and stained with a VP1-specific monoclonal antibody, followed by an FITC-conjugated secondary antibody. FITC, fluorescein isothiocyanate.
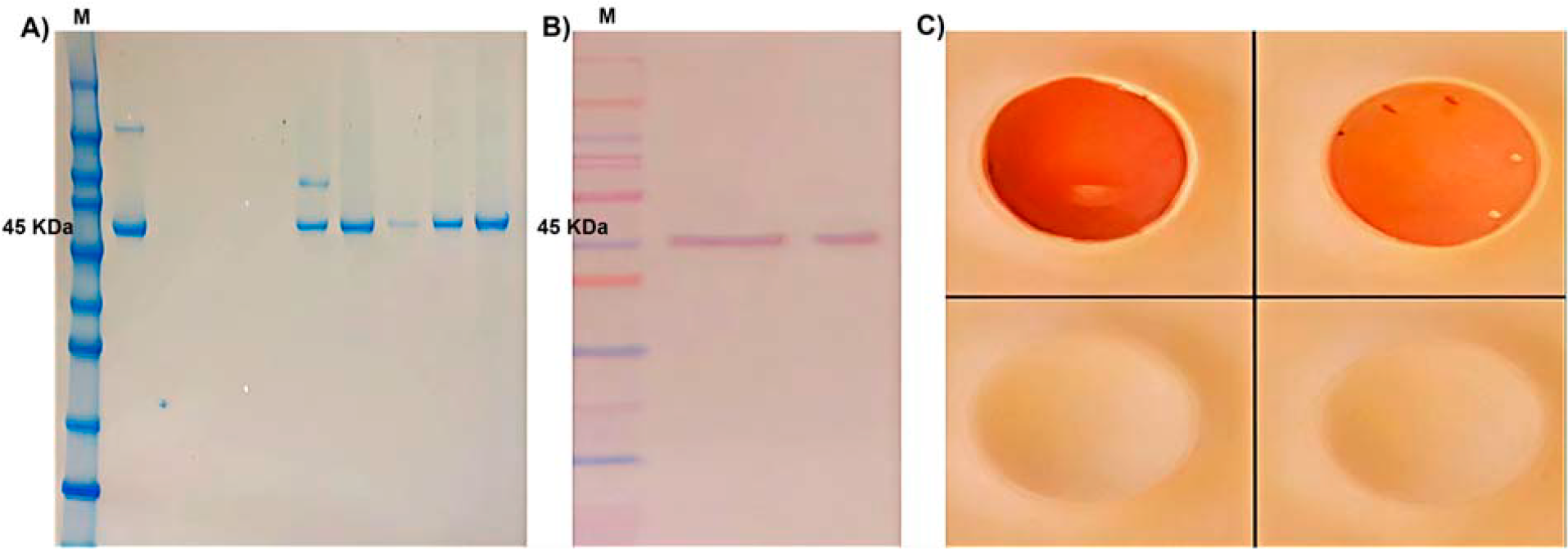
Immunochemical confirmation of recombinant BK polyomavirus (BKPyV) VP1 expression and antigenicity in Sf9 insect cells.
Exploratory Assessment of VP1 Expression Feasibility in Sf9 Cells Using Plasmid-Based Transient Transfection versus Baculovirus-Mediated Expression

The plasmid-based approach was evaluated as a feasibility experiment and produced insufficient VP1 for downstream immunoassay development.
Recombinant BKPyV VP1 protein expressed in Sf9 insect cells was purified from baculovirus-infected cell lysates using Ni–NTA affinity chromatography. Coomassie-stained SDS-PAGE analysis of the eluted fractions revealed a predominant protein band at approximately 45 kDa with minimal detectable contaminating proteins, indicating a high degree of purity. Western blot analysis using a VP1-specific monoclonal antibody confirmed immunoreactive detection of VP1 at the expected molecular weight, thereby verifying the identity of the purified antigen. Protein concentration of the purified VP1 preparation was determined by UV absorbance at 280 nm using buffer-only blanks and the theoretical extinction coefficient of VP1. Under the described expression and purification conditions, the VP1-containing eluate yielded an average concentration of approximately 1.55 mg/mL from a representative 10 mL Sf9 insect cell culture containing approximately 1 × 107 cells. This yield was sufficient to support downstream immunochemical applications, including enzyme-linked immunosorbent assay (ELISA) development and analytical validation.
Purified recombinant VP1 was subsequently used to develop an indirect ELISA for the detection of anti-BKPyV IgG antibodies in human serum. Antigen coating concentrations were optimized by checkerboard titration, and a final coating concentration of 200 ng per well was selected based on the optimal signal-to-background ratio obtained with PCR-confirmed positive and negative control sera. Under these conditions, measurable antibody reactivity above background (OD405 > 0.2) was detectable with as little as 25 ng per well of VP1 when tested using BKPyV-positive sera diluted 1:40. The optimized assay conditions included a serum dilution of 1:40 and a 1:4000 dilution of HRP-conjugated anti-human IgG secondary antibody. Each assay plate incorporated internal quality controls consisting of duplicate positive and negative control sera, along with blank wells. Samples yielding OD values at or near the predefined cutoff were retested in duplicate to minimize the risk of misclassification.
Analytical performance of the ELISA was evaluated using a PCR-defined validation panel comprising 30 serum samples (15 BKPyV-positive and 15 BKPyV-negative). Optical density (OD405) value distributions were visualized using box-and-whisker plots (Fig. 4). BKPyV-positive sera exhibited markedly higher OD values (median = 1.40) compared with PCR-negative controls (median = 0.27), demonstrating clear discrimination between the two groups.
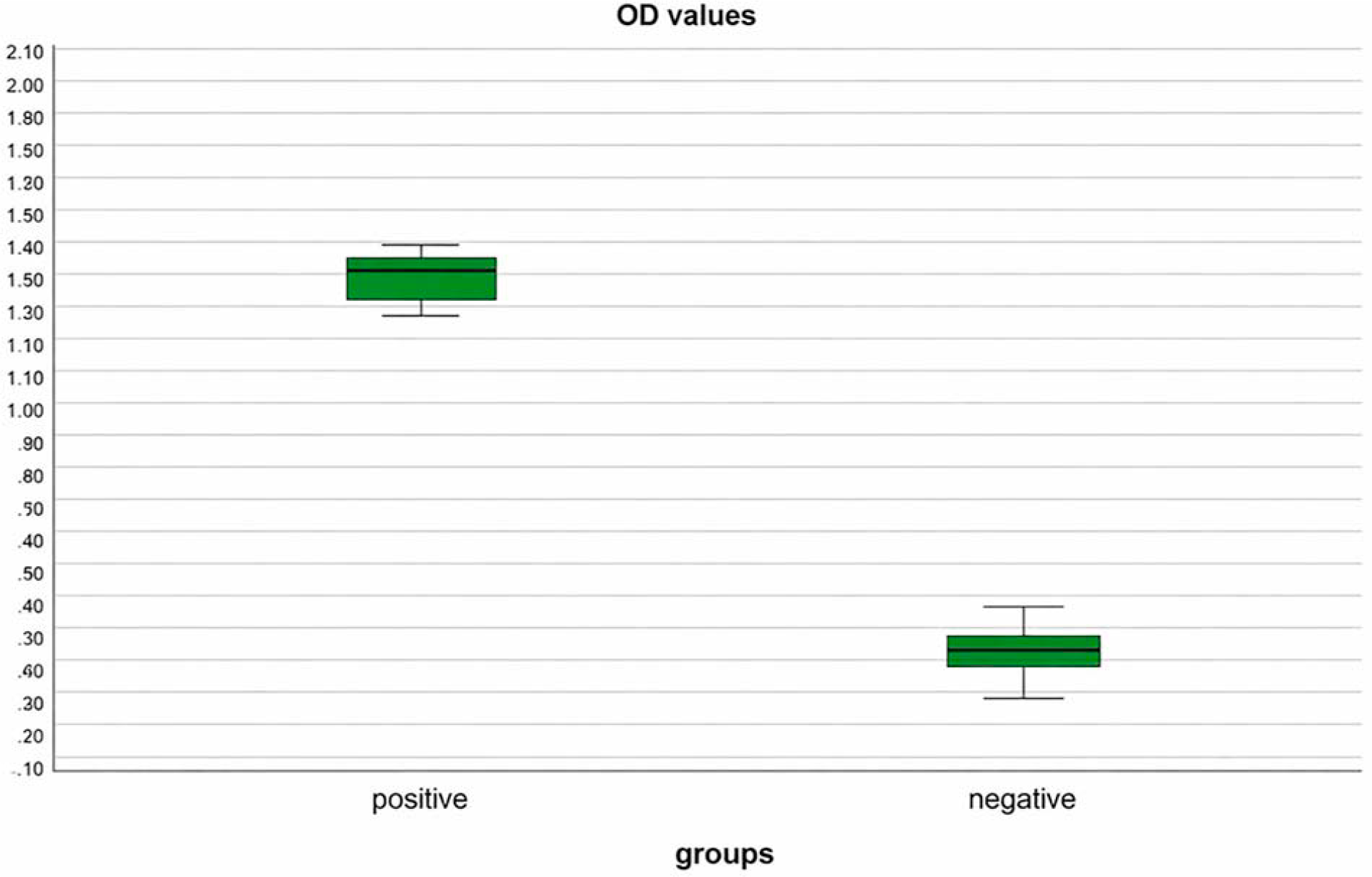
Distribution of ELISA optical density (OD405) values obtained using the recombinant BKPyV VP1-based indirect ELISA in a PCR-defined analytical serum panel (n = 30). Serum samples are grouped as BKPyV-positive (n = 15) and PCR-negative controls (n = 15). Box-and-whisker plots represent the median and interquartile range, with whiskers indicating the minimum and maximum OD values observed for each group.
Receiver operating characteristic (ROC) curve analysis was performed using mean OD405 values derived from triplicate measurements of the same PCR-defined panel to assess the analytical discriminatory performance of the VP1-based ELISA (Fig. 4). The AUC was 0.996 (95% CI: 0.973–1.000), indicating very good separation of test groups in this limited panel. Confidence intervals were wide and results should be regarded as preliminary analytical validation rather than definitive diagnostic accuracy estimates. Using the optimal cutoff determined by the Youden index (OD405 = 1.29), the assay achieved a sensitivity of 93.3% and a specificity of 100%. This ROC-derived cutoff was applied exclusively for analytical performance assessment and was not used for seroreactivity classification in the epidemiological cohort. Given the modest sample size, the ROC analysis is presented as an exploratory evaluation rather than a definitive diagnostic validation. The PCR-defined validation panel served as the analytical reference standard for ROC construction, whereas the larger cohort was analyzed using a fixed seroreactivity cutoff derived from negative controls (Fig. 5).
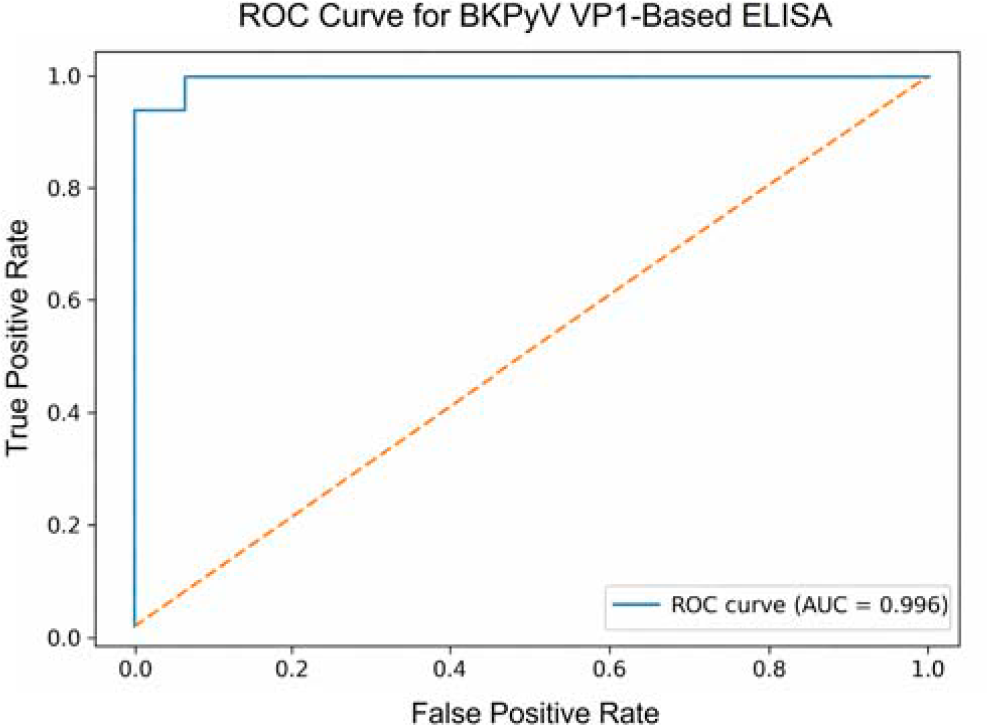
Receiver operating characteristic (ROC) curve analysis of the recombinant BKPyV VP1-based indirect ELISA using a PCR-defined analytical serum panel (n = 30; 15 BKPyV-positive and 15 BKPyV-negative sera). ROC analysis was performed using mean OD405 values obtained from triplicate measurements. The assay demonstrated very good analytical discrimination in this small validation panel, with an area under the curve (AUC) of 0.996.
To define a threshold for distinguishing seroreactive from nonreactive samples, the ELISA cutoff value was calculated using PCR-negative control sera. The cutoff was defined as the mean OD405 of the negative control group plus three standard deviations (mean + 3SD), a conservative approach commonly applied to maximize specificity in serological assays. This calculation was based on OD measurements obtained from 15 PCR-negative serum samples tested under identical assay conditions.
The negative control group exhibited a mean OD of 0.27 with a standard deviation of 0.09, resulting in a calculated cutoff value of 0.54 (Table 3). The intra-assay coefficient of variation (CV) for negative controls was 33.3%, reflecting increased relative variability at low absorbance values, a phenomenon frequently observed near background signal levels. Given this variability, samples yielding OD values at or near the cutoff were classified as borderline and retested in duplicate to improve classification consistency.
ELISA Optical Density Parameters for PCR-Defined BKPyV-Negative and BKPyV-Positive Control Serum Samples (n = 15 per Group)
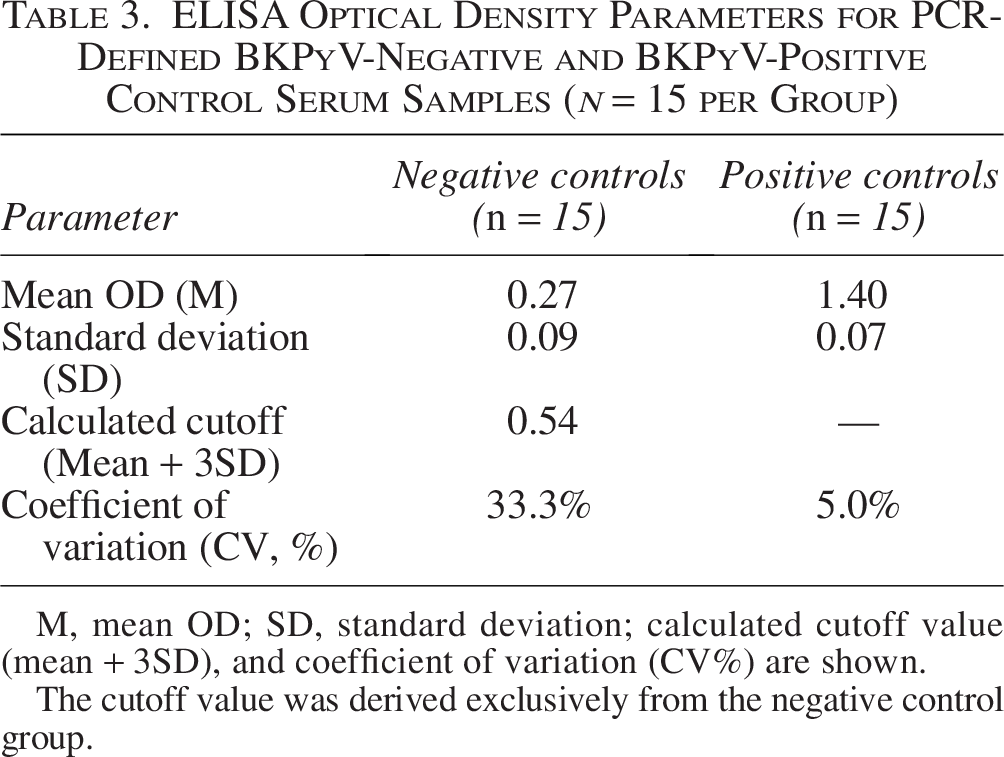
M, mean OD; SD, standard deviation; calculated cutoff value (mean + 3SD), and coefficient of variation (CV%) are shown.
The cutoff value was derived exclusively from the negative control group.
A total of 149 de-identified human serum samples spanning an age range of 1–60 years were analyzed for anti-BKPyV IgG reactivity using the validated VP1-based ELISA. Each sample was tested in duplicate, and borderline samples were retested to ensure classification accuracy. All assay plates included internal quality controls consisting of duplicate positive and negative control sera and blank wells.
Within this convenience cohort, 89.7% of samples exhibited ELISA reactivity above the predefined cutoff (Table 4). Nonreactive samples displayed consistently low OD values (mean OD 0.27), whereas reactive samples showed substantially higher OD values (median 1.40). The difference in OD values between reactive and nonreactive groups was statistically significant (Mann–Whitney U test, p < 0.0001).
Distribution of BKPyV VP1-Based ELISA Reactivity across Age Groups, Gender, and Clinical Categories

To explore age-associated patterns of antibody reactivity, mean OD405 values were plotted across predefined age groups (Fig. 6). A stepwise increase in IgG reactivity with age was observed, with higher mean OD values in older age groups. Detectable IgG reactivity was also observed in a subset of children under 5 years of age; however, given the cross-sectional study design, this finding may reflect early-life exposure, passive maternal IgG transfer, or a combination of both and should therefore be interpreted cautiously.
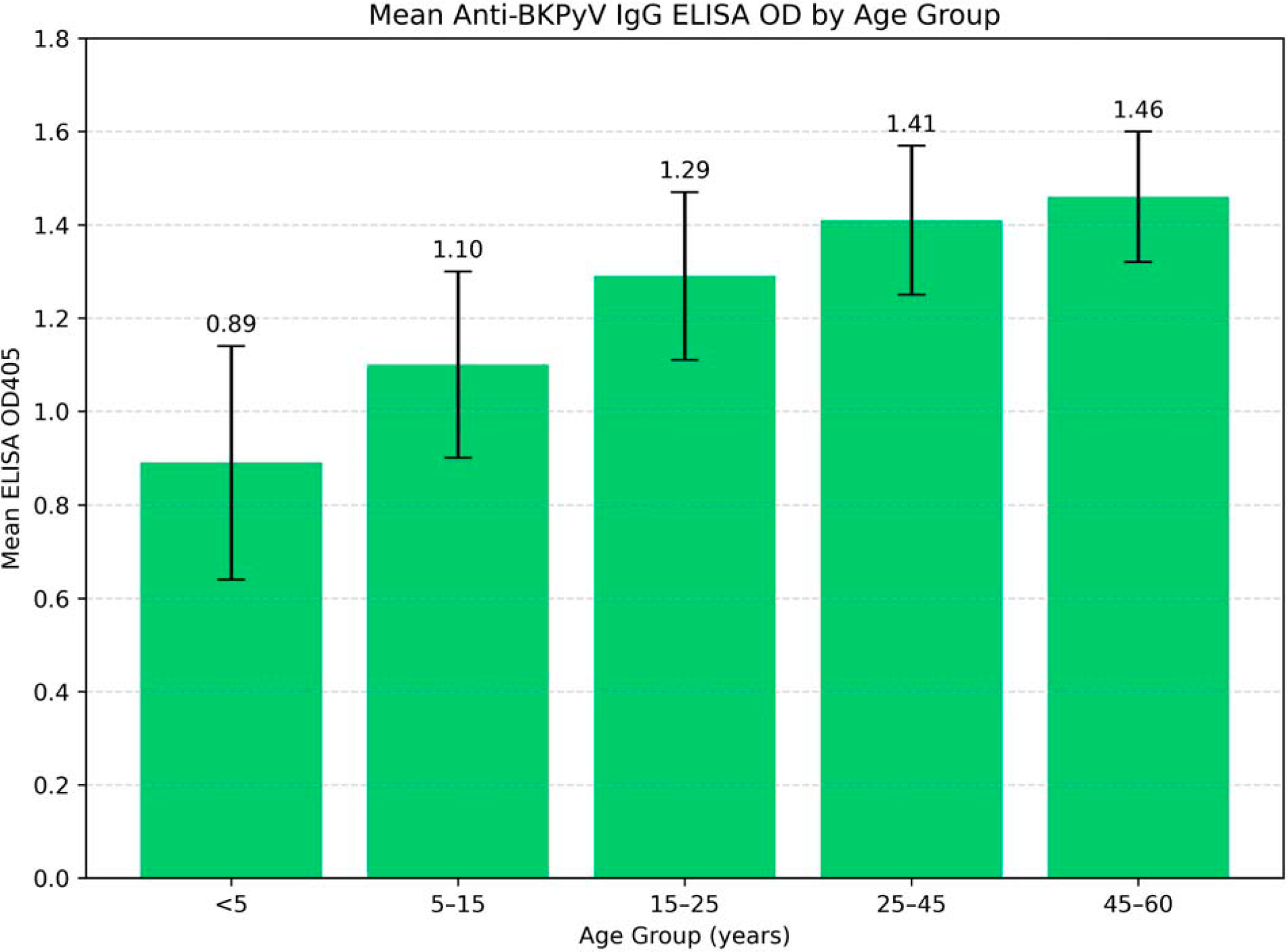
Mean anti-BK polyomavirus (BKPyV) IgG ELISA OD405 values across age groups in the analyzed cohort. Bars represent mean OD values obtained using the VP1-based indirect ELISA, and error bars indicate the standard deviation within each age group.
Reactivity patterns were further examined according to gender and clinical status (Table 4). No significant difference in mean OD values was observed between female and male participants. Higher mean OD values were observed among renal transplant and hemodialysis patients compared with healthy individuals (p < 0.001), whereas HIV-positive individuals showed intermediate levels of reactivity. Given the nonrandomized sampling strategy and limited demographic scope, these findings describe serological reactivity patterns within the analyzed cohort rather than population-level prevalence estimates.
Discussion
This study presents a comprehensive evaluation of BKPyV VP1 expression using an insect cell-based baculovirus system and its application in the development of a serological ELISA, addressing an important diagnostic and epidemiological gap in BKPyV surveillance (Touzé et al., 2001; Husseiny and Lacey, 2011; Cook, 2016). The results demonstrate that baculovirus-mediated expression enables the production of high-yield, immunoreactive VP1 antigen suitable for analytical assay development, while also providing insight into population-level seroreactivity patterns across age and clinical subgroups (Leavitt et al., 1985; Gillock et al., 1997; Noteborn et al., 1998; Goldmann et al., 1999; Lee et al., 2011; Zhao et al., 2019). A key limitation of the present work is that higher-order assembly of VP1 into pentamers or virus-like particles was not directly evaluated. Therefore, antigenic authenticity is inferred from serological reactivity rather than confirmed structurally. Future work should incorporate native PAGE, transmission electron microscopy (TEM), dynamic light scattering (DLS) or cryo-electron microscopy to verify VP1 assembly status. Accordingly, no claims are made regarding the presence of fully native quaternary structures, and the antigen used here should be regarded as structurally unverified at this stage.
The recombinant baculovirus system yielded approximately 1.55 mg/mL of purified VP1 from a 10 mL Sf9 culture, which is broadly consistent with yields reported for baculovirus-based systems, although direct comparisons are limited by methodological heterogeneity (Viscidi et al., 2003; Hong et al., 2022). Although expression kinetics were not formally evaluated over multiple time points, the achieved yield and purity were sufficient for downstream immunochemical applications. Importantly, while a plasmid-based transient expression strategy was explored, it produced negligible VP1 levels and was therefore retained only as a feasibility assessment. These findings support reframing the study as a validation of baculovirus-mediated expression rather than a direct comparative analysis, consistent with the limitations of transient plasmid systems for diagnostic-grade antigen production (Abedi Kiasari, 2020; Lampinen et al., 2024).
The use of a His-tag enabled single-step Ni–NTA purification, yielding VP1 with >95% apparent purity and no detectable degradation products by SDS-PAGE and immunoblotting. However, this study did not directly assess higher-order VP1 assembly into pentamers or VLPs (Vassylyeva et al., 2017). Since conformational epitope preservation is critical for serological authenticity, this represents an important limitation. Prior studies have demonstrated that VP1 expressed in insect cells can self-assemble into pentameric structures or VLPs; therefore, future investigations should incorporate native PAGE, TEM, or DLS to confirm structural organization and further strengthen antigen characterization (Li et al., 2003; Hurdiss et al., 2016). Until such analyses are performed, the presence of conformational epitopes should be considered probable but not definitively demonstrated.
The VP1-based ELISA demonstrated strong analytical discrimination between PCR-confirmed BKPyV-positive and negative sera. Using a PCR-defined validation panel (n = 30), the assay achieved an estimated sensitivity of 93.3% and specificity of 100% at the ROC-derived optimal cutoff. ROC analysis yielded an AUC of 0.996 (95% CI: 0.973–1.000), showing clear separation of positive and negative groups in this dataset. However, these estimates should be interpreted with caution given the small size of the PCR-defined validation cohort (n = 30), which limits the precision of sensitivity and specificity estimates and contributes to wide confidence intervals. Accordingly, the current findings reflect exploratory analytical validation rather than definitive diagnostic accuracy, and independent external validation in larger cohorts will be essential before clinical application is considered. The relatively high CV among negative controls (33%) likely reflects low OD values close to background levels and may affect reproducibility near the cutoff; future optimization should therefore include additional quality-control measures and inter-assay variability assessment.
The conventional cutoff derived from the mean plus three standard deviations of negative controls (OD405 = 0.54) was intentionally conservative to maximize specificity for epidemiological screening. The ROC-derived cutoff was used exclusively for analytical performance evaluation and not for seroreactivity classification in the population cohort. This approach is particularly important in assays where variability is higher near the cutoff, as observed here. Together, these approaches highlight the importance of incorporating both distribution-based and ROC-based methods, and support the future introduction of a “gray zone” or equivocal category for borderline sera, particularly in large-scale validation studies.
Application of the validated ELISA to a convenience cohort of 149 sera showed an overall seroreactivity rate of 89.7%, consistent with previous reports of high BKPyV seroprevalence in adults (Kean et al., 2009; Gossai et al., 2016; Blackard et al., 2020). Antibody reactivity increased progressively with age, supporting the notion of early-life exposure followed by persistent or intermittently boosted humoral immunity. Detectable IgG reactivity was also observed in a subset of children under 5 years of age. Because of the cross-sectional design, this finding should be interpreted cautiously, as it may reflect either early primary infection or passive transplacental transfer of maternal IgG, or a combination of both. The absence of IgM testing and IgG avidity assessment prevents clear differentiation between these possibilities; future studies incorporating these methods will be required to distinguish passive immunity from true primary infection in pediatric populations (Wang et al., 2020).
Higher mean OD values were observed in renal transplant and hemodialysis patients compared with healthy individuals, consistent with frequent BKPyV reactivation under immunosuppression. However, the study did not include longitudinal sampling before and after transplantation or during clinical reactivation episodes. As a result, the present findings describe serological status rather than dynamic immune responses. Longitudinal studies will be essential to determine whether VP1 IgG levels, subclasses, or avidity changes can inform clinical monitoring or risk stratification in high-risk populations.
The seropositivity rate observed among healthy adults was lower than expected, likely reflecting sampling bias related to non-randomized recruitment and limited demographic diversity. Accordingly, the epidemiological findings should not be interpreted as population-level prevalence estimates. Multicenter studies with stratified sampling and larger cohorts will be required to improve representativeness and external validity.
Finally, although VP1-based serology is widely used, potential cross-reactivity with other human polyomaviruses, including JCPyV and SV40, remains an important consideration. Structural homology among VP1 proteins may lead to partial serological cross-reactivity, which was not directly assessed in the present study. Future validation using genotype-specific antigens or competitive inhibition assays will be necessary to fully define assay specificity (Viscidi and Clayman, 2006; Kean et al., 2009; Moens et al., 2013).
In summary, this study demonstrates that baculovirus-expressed BKPyV VP1 is a robust and scalable antigen for serological assay development. Despite limitations related to structural characterization, sample size, and cross-reactivity assessment, the developed ELISA shows promising analytical performance in a limited validation panel and provides a solid foundation for expanded validation, epidemiological surveillance, and potential clinical application in immunocompromised populations (Hirsch et al., 2013; Kotton et al., 2024). These results should therefore be viewed as preliminary and hypothesis-generating rather than confirmatory.
Conclusion
In conclusion, this study demonstrates that baculovirus-mediated expression of BKPyV VP1 in insect cells provides a robust and scalable source of recombinant antigen with high purity and preserved immunoreactivity. The VP1 protein produced under these conditions supported the development of an indirect ELISA capable of reliably discriminating BKPyV-seropositive and seronegative sera, showing promising analytical performance in a small PCR-defined validation panel. Although analytical validation was conducted on a limited number of reference samples, the assay performance metrics and the clear separation of OD values support the potential suitability of this platform for serological applications. Application of the VP1-based ELISA to a broader convenience cohort revealed age-dependent seroreactivity patterns and elevated antibody levels in immunocompromised individuals, findings that are consistent with known BKPyV epidemiology. Several limitations should be acknowledged, including the absence of direct structural characterization of VP1 assembly, the modest size of the analytical validation cohort, and the lack of assessment of potential cross-reactivity with other human polyomaviruses. Future studies incorporating structural analyses, larger multicenter validation panels, and longitudinal sampling in high-risk populations will be essential to further define the diagnostic and clinical utility of this assay. Overall, the baculovirus-expressed VP1 antigen and the associated ELISA described here provide a solid foundation for expanded seroepidemiological studies and represent a practical platform for further development of BKPyV serological tools in both research and clinical contexts. These findings should be regarded as preliminary and require confirmation in independent cohorts before routine clinical implementation is considered.
Authors’ Contributions
N.H.: and B.A.K. Conceptualization. N.H.: Methodology, validation, investigation, data curation, and writing—original draft. B.A.K.: Resources, writing—review and editing, supervision, project administration, and funding acquisition. M.H.: Formal analysis and visualization.
Footnotes
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
No funding was received for this article.