
Abstract
Noroviruses are single-stranded positive-polarity RNA viruses classified in the family Caliciviridae. Human noroviruses (HuNoVs) are a leading cause of acute viral gastroenteritis worldwide. Despite their ability to infect various epithelial and nonepithelial cell types, establishing robust in vitro culture systems for HuNoVs remains challenging. As a result, murine norovirus 1 (MNV-1) has become a widely used model for investigating norovirus biology and pathogenesis, due to its ability to replicate efficiently in primary dendritic cells and macrophages. Although different B-cell lines are susceptible to MNV-1 infection, the susceptibility of primary B lymphocytes has been poorly characterized. Here, we demonstrate that MNV-1 infects primary B lymphocytes, with infection levels increased by prior stimulation with lipopolysaccharide and interleukin-4. The enhanced infection does not appear to result from increased virion binding but instead to an increased cellular permissiveness. These findings provide new insights into the cellular tropism of MNV-1 and suggest that the activation status of B-cell influences their susceptibility to infection. This model may improve our understanding of norovirus–host-cell interactions in adaptive immune cells and could aid in the development of more representative in vitro systems for studying norovirus pathogenesis.
Introduction
The family Caliciviridae comprises viruses that cause various diseases in different vertebrates. Human noroviruses (HuNoVs) are the etiologic agents responsible for acute gastroenteritis (Patel et al., 2008; Widdowson et al., 2005). Historically, the propagation of HuNoV has been challenging; however, feline calicivirus (FCV) and murine noroviruses (MNVs), which belong to the same genus as HuNoV, serve as valuable models for studying the biology and pathogenesis of members of this family (Wobus et al., 2006).
MNV-1 has a tropism for multiple epithelial and nonepithelial cell types, including tuft cells (Wilen et al., 2018), macrophages, and dendritic cells (Wobus et al., 2004), and B lymphocytes (Jones et al., 2014). Notably, B lymphocytes are a common target for both HuNoV and MNV (Jones et al., 2014). Different B-cell cultures are permissive to MNV infection (Jones et al., 2014), and viral proteins and RNA have been found in B lymphocytes of Peyer’s patches in mice (Jones et al., 2014). Although there is evidence that B lymphocytes are permissive to MNV-1 infection, this model is not as efficient as others, such as the macrophage RAW 264.7, a murine cell line derived rom an Abelson murine leukemia virus-induced tumor (Jones et al., 2014). However, the ability of B lymphocytes to become infected with HuNoV represents an advantage over other models, highlighting the need to develop strategies to strengthen and optimize this infection model.
Here, we demonstrate that stimulation of B lymphocytes with lipopolysaccharide (LPS) and interleukin-4 (IL-4) does not affect virus binding but enhances MNV infection, leading to increased production of nonstructural proteins and viral particles. These findings suggest that immune activation improves the susceptibility of B lymphocytes to MNV and may help establish a more robust and physiologically relevant in vitro model for studying norovirus biology.
Materials and Methods
Mice
Eight- to ten-week-old male C57BL/6J mice were bred and maintained at the UPEAL-Cinvestav-IPN facilities (Mexico City, Mexico). Mice were maintained on a 12-h light/dark cycle. All experiments involving mice were conducted according to relevant guidelines and regulations and were approved by the Cinvestav-IPN Animal Care and Use Committee (145–15).
Cell cultures and viruses
The RAW 264.7 cell line was obtained from the American Type Culture Collection (ATCC) (Rockville, MD) and maintained in Dulbecco’s modified Eagle’s medium-high glucose medium (Gibco BRL Cat. No. 12100-046) supplemented with 10% fetal bovine serum (FBS; HyClone Standard, Cat. No. SH30088.03) and 1× Pen/Strep (Gibco, Cat. No. 15140-122). For the isolation of primary B lymphocytes, mononuclear splenic cells were obtained using a Ficoll-Paque Premium gradient (GE Healthcare Cytiva, Cat. No. 17-5442-03), and B220+ cells were enriched by negative selection with anti-Thy-1.1 (NIM-R1) monoclonal antibody ascites-coated plastic plates (Chayen and Parkhouse, 1982). The obtained B lymphocytes were incubated at 37°C and 5% CO2 in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco Cat. No. 31800-089) supplemented with 10% fetal bovine serum and 1× Pen/Strep. B-cell stimulation was induced by incubation with LPS from Escherichia coli O55:B5 (20 μg/mL) (Sigma-Aldrich, Cat. No. F8666) plus the addition of IL-4 (10 U/mL) (R&D Systems, Cat. No. 204-IL) for 48 h at 37°C in 5% CO2.
MNV-1 was purchased from ATCC and propagated in RAW 264.7 cells as previously reported (Bailey et al., 2010).
Lymphocyte infection
A total of 2 × 106 B lymphocytes were cultured in 500 µL of RPMI 1640 medium and incubated with MNV-1 at a multiplicity of infection (MOI) of 5 for 1 h, under orbital shaking (150 rpm; 3-mm orbit) at 37°C. B lymphocytes were washed twice with sterile phosphate-buffered saline (PBS; pH 7.4), suspended in 1 mL of RPMI 1640 medium supplemented with 10% FBS, and incubated for 24 h at 37°C in 5% CO2. B lymphocytes were sedimented by centrifugation at 300 g for 5 min, and the supernatant was separated to subsequently quantify the viral titer by plaque assay in RAW 264.7 cells. For viral protein detection, the cell pellet was lysed using SDS lysis buffer (25 mM HEPES, 2 mM EDTA, 25 mM NaF, 1% SDS, pH = 7.6) with 1× SIGMAFAST™ Protease Inhibitor Cocktail EDTA-Free (SIGMA Cat. No. S8830); samples were sonicated using three cycles of 10 sec and 40% amplitude with 15 sec of break and analyzed by Western blotting.
Antibodies
PE or Pacific Blue anti-B220/CD45R (clone RA3-6B2; BioLegend, Cat. No. 103207, 103227); APC goat (minimal cross-reactivity) anti-rat IgG1 (Poly 4054; BioLegend, Cat. No. 405407); Alexa fluor 488 (AF488) donkey anti-rabbit IgG (H + L) (Invitrogen, Thermo Fisher Scientific, Cat. No. A21206); rabbit IgG anti-CD300lf (MyBiosource); anti-mouse IgG 680LT (LI COR Biosciences, Cat. No. 926-68020), anti-rabbit IgG800CW (LI COR Biosciences,Cat. No. 926-32211), and anti-rat IgG 800CW (LI COR Biosciences, Cat. No. 926-32219); Peroxidase AffiniPure™ goat anti-rabbit IgG (H + L) (Jackson Immuno Research, Cat. No. 111-035-003); anti-CD44 hybridoma culture supernatant ( clone IM7); mouse IgG anti-actin hybridoma supernatant (kindly donated by Dr. Jose Manuel Hernandez, CINVESTAV, Mexico); and anti-MNV NS7 protein (kindly donated by Dr. Ian Goodfellow, University of Cambridge, UK).
Binding assay
B lymphocytes were incubated with MNV-1 at an MOI of 100 for 30 min at 4°C, and the cells were washed twice with cold PBS to remove the unbound virus. The cell pellet was stored at −80°C until it was used. The RNA extraction was performed with a BioPure RNA Isolation Kit (Cat. No. RNAv-090). cDNA was obtained using the LunaScript RT SuperMix Kit (New England Biolabs, Cat. No. E3010L) according to the manufacturer’s recommendations, with 500 ng of RNA used. The qPCR assay was performed using Luna Universal Probe qPCR master mix (New England Biolabs Cat. No. M3003E). As a control, a region of actin was amplified with the sense primer 5′-CCTCTATGCCAACACAGTGC-3′, antisense primer: 5′-GCTAGGAGCCAGAGCAGTAA-3′, and probe 5′-HEX-ACCCAGGCATTGCTGACAGGATGCA-BHQ-1–3′. RT-PCR was performed on the Bio-Rad CFX96 Touch Real-Time PCR Detection System under the following conditions: one cycle at 95°C for 1 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 30 sec, with a plate read. Relative expression was performed using Bio-Rad CFX Manager software, which served as a control for expression.
Plaque assay
For virus quantification, 24-well multiplates were used in which approximately 1 × 105 RAW 264.7 cells per well were seeded in Dulbecco’s modified Eagle’s medium-high glucose medium supplemented with 10% FBS. The assay was performed when the cells reached 80% confluency. Four serial 10-base dilutions (1:10) were prepared in DMEM medium from the supernatants obtained from infected B lymphocytes. After removing the medium, 300 µL of each dilution were carefully added in duplicate to the cell monolayers. The plates were incubated for 1 h at 37°C. Then, the supernatant was removed, and 500 µL of 1% carboxymethylcellulose (MERCK Millipore Cat. No. 419303) solution in DMEM medium and 2% FBS were added. The plates were incubated for 30–36 h at 37°C. After this time, the medium was removed from each well, and 300 µL of 3.7% formaldehyde (REPROQUIFIN Cat. No. JTB-2106-02) was placed and incubated for 15 min at room temperature. The formaldehyde was removed, and 300 µL of crystal violet solution was added. Then, the plates were incubated for 10 min and revealed by immersion in a container with water. The plates were allowed to dry, and the number of lytic plaques was counted.
Western blot assay
Thirty micrograms of protein extract were mixed with 6× Laemmli buffer (10% Tris, 1 M pH 6.8, 1% SDS, 10% glycerol, 0.1% β-mercaptoethanol) and subjected to electrophoresis in a 10% acrylamide gel in Tris-glycine buffer at 80 V. The proteins were transferred to a nitrocellulose membrane 0.2 µm (BIO-RAD Cat. No. 1620112) for 1.5 h in a humid chamber in Tris-glycine buffer and 20% methanol; the membrane was blocked for 1 h with Odyssey® Blocking Buffer solution (PBS) (ODYSSEY Infrared Imaging System Cat. No. 927-40000) and incubated with a polyclonal rabbit anti-NS7 antibody (1:1,000 dilution in a 5% skim milk/TBS-Tween) overnight at 4°C under agitation. The membrane was washed twice with TBS-Tween and incubated with the secondary antibody (peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG [H + L], Jackson ImmunoResearch, at a 1:10,000 dilution in TBS-Tween) for 1 h at room temperature under constant agitation. Finally, three additional washes with TBS-Tween were performed. The membrane was incubated with SuperSignal West Femto substrate (Thermo Fisher Scientific, Cat. No. 34094), and images were acquired using the ChemiDoc Imaging System.
Results
Mouse spleen B lymphocytes are permissive to infection with MNV-1
To determine if primary B lymphocytes were susceptible to MNV-1 infection, purified murine B lymphocytes from the spleen were obtained by panning negative selection with an anti-Thy1 antibody, and a viral binding assay was performed (Fig. 1A). Then, the purified B lymphocytes were used to test the ability of MNV-1 to bind and enter; thus, B lymphocytes were mock-infected or infected with MNV-1 at an MOI of 100 for 30 min at 4°C to ensure saturation of viral binding under nonpermissive conditions for viral entry, and RT-PCR quantified the bound virus. The relative expression was determined using the CFX manager software (Fig. 1A). Actin was used as an expression control. We found the presence of the MNV-1 viral genome in B lymphocytes incubated with MNV-1, suggesting that this virus can bind to primary B lymphocytes from the murine spleen.

Spleen B lymphocytes are permissive to MNV-1 infection.
To determine whether murine splenic B lymphocytes support productive MNV-1 infection, cells were mock-infected or exposed to either untreated or UV-inactivated MNV-1 at an MOI of 5 for 24 h; a lower MOI was used to evaluate productive infection and viral protein synthesis under conditions permissive for replication. The viral NS7, corresponding to the RNA-dependent RNA polymerase, was assessed in total protein lysates using Western blotting. Protein extracts from RAW 264.7 macrophages infected with MNV-1 were used as a positive control (Fig. 1B). NS7 protein was detected in MNV-1-infected primary B lymphocytes, although its expression was approximately 5.6-fold lower than in infected RAW 264.7 cells (Fig. 1B and C). In contrast, NS7 was not detected in lysates from mock-infected B lymphocytes and RAW 264.7 cells or those exposed to UV-inactivated MNV-1, indicating that MNV-1 is capable of infecting murine splenic primary B lymphocytes and initiating viral protein synthesis.
Activation of murine splenic B lymphocytes favors MNV-1 infection
Activation of B lymphocytes enhances cell viability and proliferation, alters the cell cycle state, and promotes the upregulation of surface molecules relevant to an infection (Kehrl et al., 1984b; Xu et al., 2008). Therefore, activating B lymphocytes could enhance the efficiency of specific components of the replicative machinery, facilitating efficient viral replication.
To evaluate whether the activation of B lymphocytes favors MNV-1 infection, primary B cells were stimulated or not with LPS + IL-4 for 48 h, as this time point corresponds to the peak activation of CD44 (gp90), previously determined by our workgroup (SANTOS‐ARGUMEDO et al., 1997). The activation of B lymphocytes was verified by measuring CD44 expression on their surface using flow cytometry (Fig. 2A). B lymphocytes were mock-infected or exposed to either untreated or UV-inactivated MNV-1 at an MOI of 5 for 24 h, and the amount of the viral protein NS7 was assessed by Western blotting (Fig. 2B). B lymphocyte activation caused an increase of approximately three times the NS7 viral protein levels compared to the unstimulated lymphocytes (Fig. 2B and C), indicating that the activation of B lymphocytes favors NS7 production.
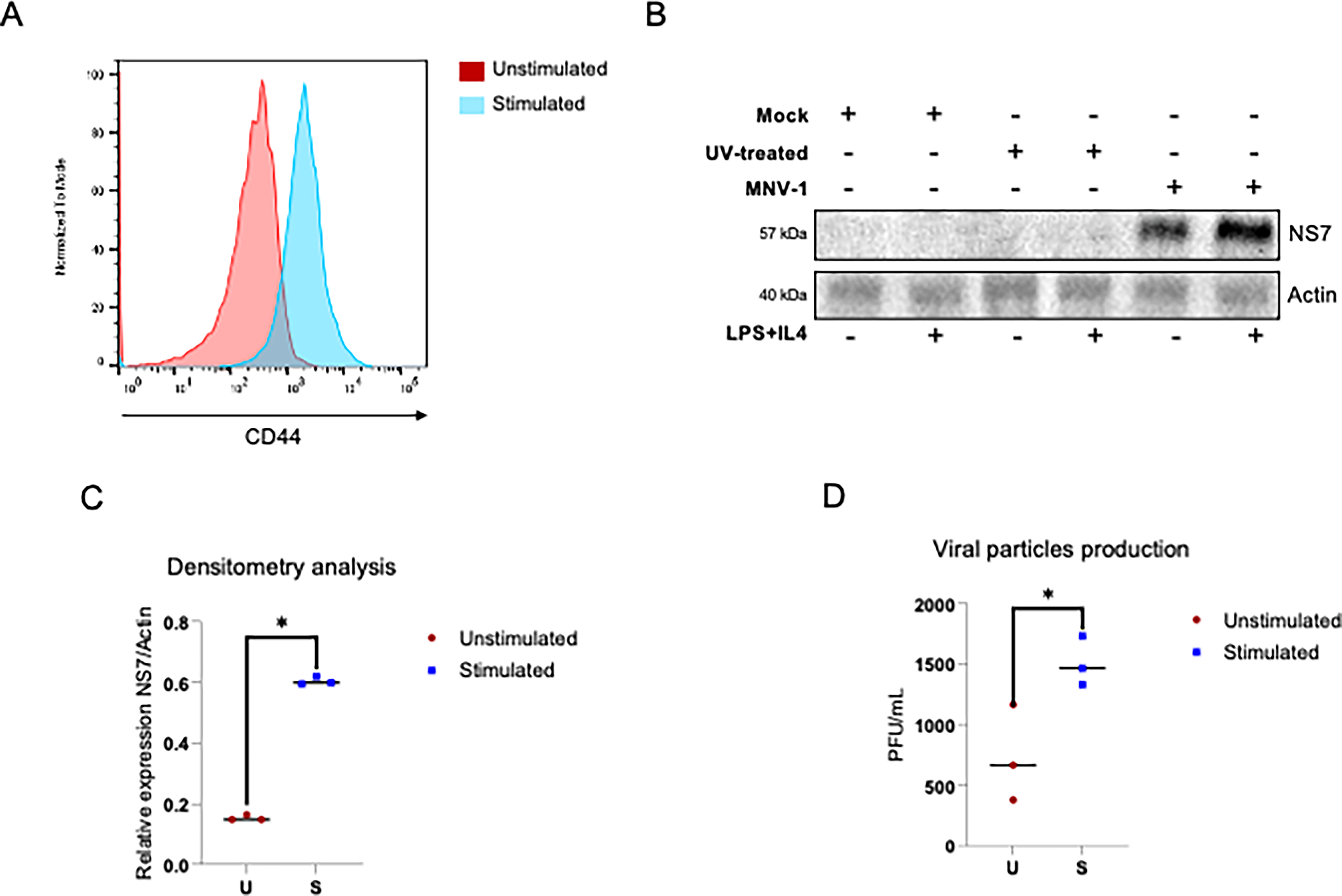
B lymphocyte activation favors MNV-1 infection and virus yield.
To determine whether this increased susceptibility to infection translates into major production of viral particles, the infective viral particles in the supernatant-associated fractions from infected stimulated B lymphocytes were quantified and compared to those in unstimulated B lymphocytes by plaque assays in RAW 264.7 cells (Fig. 2D). The viral particle production in the supernatant-associated fractions from stimulated B lymphocytes with LPS + IL-4 and infected with MNV-1 at an MOI of 5 for 72 h was two times the amount of the viral particles from unstimulated B lymphocytes (Fig. 2D), indicating that B-lymphocyte activation favors MNV-1 replication and viral particle production.
It has been reported that the activation of B lymphocytes increases the surface expression of various molecules (Kehrl et al., 1984b, 1984a), some of which may function as receptors for MNV-1. Molecules, such as CD44, CD36, CD98, and transferrin receptors, have been described to participate in MNV-1 binding to bone marrow dendritic cells (Bragazzi Cunha and Wobus, 2016), while CD300lf is the main receptor for MNV-1 on macrophages (Haga et al., 2016). Increasing any of these molecules due to B lymphocyte activation might facilitate MNV-1 binding and entry into these cells. The expression of some molecules involved in MNV-1 binding on B lymphocytes was analyzed using data from the Gene Expression Commons (GEXC) database (https://gexc.riken.jp) (Seita et al., 2012). A heatmap was generated to compare receptor expression in two different B subsets—marginal zone and follicular zone B cells—with that in monocytes and granulocytes, which are known to express several of these receptors at high levels (Fig. 3A). We found that in murine monocytes, CD300lf, the MNV receptor (Graziano et al., 2020), and CD300ld, which is involved in MNV attachment (Haga et al., 2016), are highly expressed. In contrast, only CD300lf has been reported in B lymphocytes. Moreover, other molecules implicated in MNV attachment were analyzed: CD44 was detected in both monocytes and lymphocytes; CD36 was found only in lymphocytes; and CD98 was not reported in either cell population.
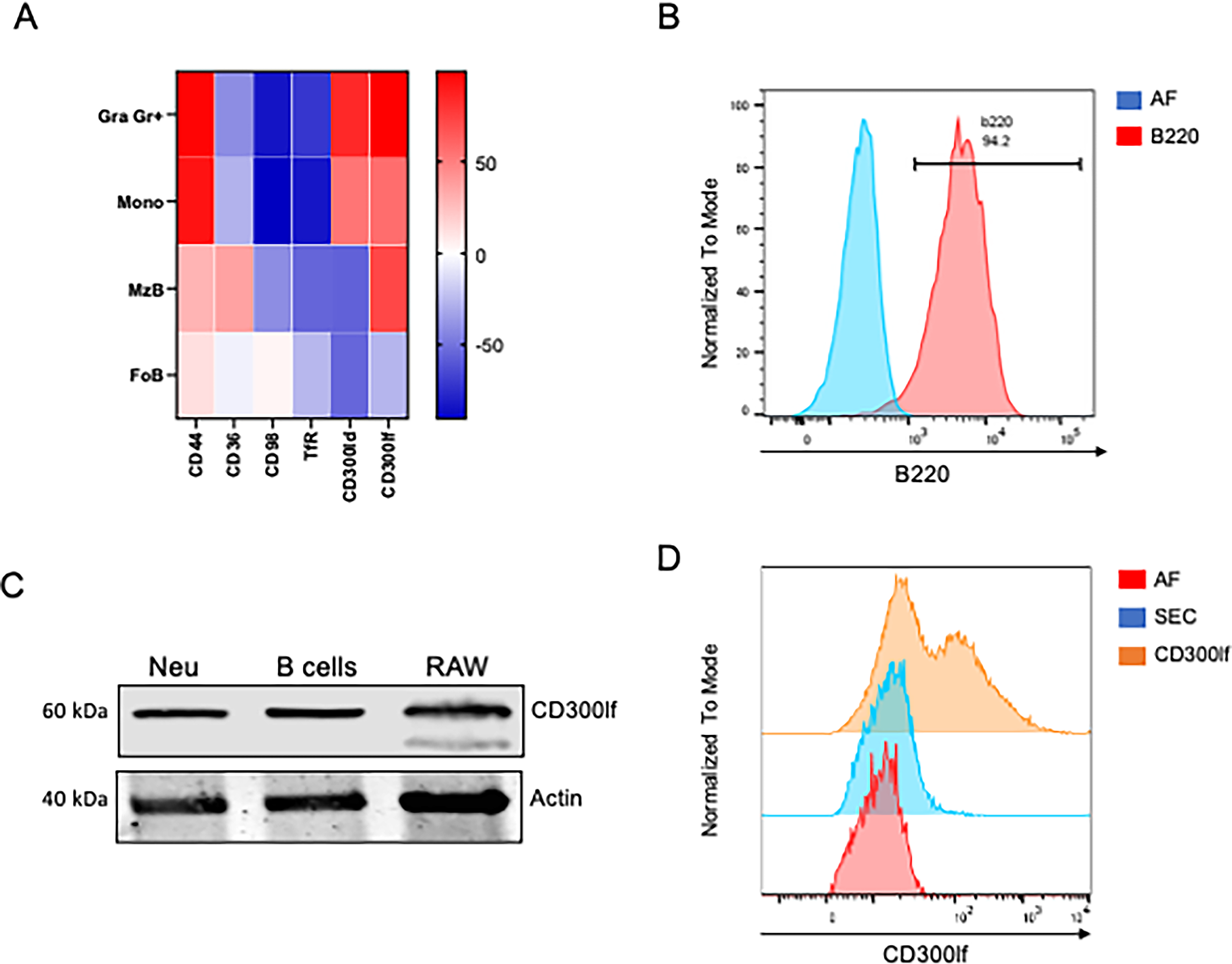
B lymphocytes express CD300lf.
To corroborate the expression of CD300lf in B lymphocytes, we purified B lymphocytes by negative selection with anti-CD90.1 panning, which captures T lymphocytes and allows B lymphocytes to remain free in the supernatant (Fig. 3B). By flow cytometry, we verified a cell population composed of approximately 95% B lymphocytes, and the expression of CD300lf was determined by Western blotting (Fig. 3C). Murine neutrophils and RAW 264.7 macrophages were used as positive controls for the presence of CD300If. The presence of a 60 kDa band, corresponding to CD300If, was detected in protein extracts from B lymphocytes, as well as from murine neutrophils and RAW 264.7 macrophages (Fig. 3C), strongly suggesting that this receptor molecule was present in B lymphocytes. These results were further corroborated by flow cytometry (Fig. 3D). B lymphocytes were selected using an anti-B220 antibody, and the CD300lf expression was evaluated with a specific anti-CD300lf antibody. Flow cytometry analysis revealed a partial shift in fluorescence intensity within the B220+ population, indicating that a subset of B lymphocytes expresses CD300lf on their surface.
Increased susceptibility of activated B lymphocytes to MNV-1 is unrelated to enhanced viral attachment
One possible explanation of the increased viral yield upon B lymphocyte activation is the upregulation of the CD300If receptor. To evaluate this possibility, primary B lymphocytes were stimulated with LPS + IL-4 for 48 h and the expression of CD300lf was measured by flow cytometry (Fig. 4A and B). CD44 expression was used to demonstrate B lymphocyte activation (Fig. 4C and D), since it can increase up to 10 times in stimulated B (Santos‐Argumedo et al., 1997). No difference in expression of CD300lf on the membrane of stimulated and unstimulated B lymphocytes was observed, compared to the sixfold increased expression of CD44 in stimulated B lymphocytes. This increase corresponds to what has been reported from our workgroup (Santos‐Argumedo et al., 1997).

Stimulation of B lymphocytes with LPS and IL-4 does not favor MNV-1 binding to B lymphocytes. Splenic B lymphocytes were isolated and stimulated (red) or not (unstimulated) (orange) with LPS (20 μg/mL) + IL-4 (10 U/mL) for 48 h. The expression of
Another possibility is that the activation of B lymphocytes may enhance the expression of other surface molecules involved in increased binding of MNV-1. To test this, unstimulated and LPS + IL-4-stimulated primary B lymphocytes were incubated with MNV-1 at an MOI of 100 for 30 min at 4°C. The amount of virus bound to the cells was quantified by RT-PCR using primers specific to ORF-1 (Fig. 4E). No significant difference was observed in the amount of virus bound to stimulated versus unstimulated B cells. These results indicate that the increased viral yield observed in activated B lymphocytes is not attributable to enhanced virus binding, as stimulation with LPS + IL-4 did not augment virus attachment to lymphocytes nor increase CD3001f expression. However, the elevated NS7 viral protein levels and the higher production of viral particles in LPS + IL-4-stimulated cells suggest that changes in post-binding events, such as viral RNA synthesis or viral protein production, may underlie the increased permissiveness of activated B lymphocytes.
Discussion
The cell tropism of noroviruses and the identity of their entry receptors have been widely discussed in the literature (Haga et al., 2016; Jones et al., 2014; Nelson et al., 2018; Wobus et al., 2004). Although significant progress has been made in both areas, many questions remain unsolved. The murine macrophage cell line RAW 264.7 has historically been a robust in vitro model for studying MNV biology and pathogenesis (Wobus et al., 2006), given its susceptibility and high-yield virus production.
Paradoxically, in vivo models show very low replication in macrophages and dendritic cells, despite the detection of viral RNA (Grau et al., 2017; Wilen et al., 2018). Tuft cells have been identified as the primary target for MNV-1 infection in mice (Wilen et al., 2018); however, they represent less than 1% of the population of epithelial cells in the intestinal tract. This low abundance of tuft cells suggests that additional permissive cell types may be involved during in vivo infection. Among them, enterocytes have been evaluated as potential MNV targets; although they do not support productive infection in vitro, they may facilitate the translocation of MNV across the intestinal barrier (Gonzalez-Hernandez et al., 2013). B lymphocytes have also been identified as targets for MNV-1, and in vitro experiments using B-cell lines have demonstrated that they are permissive to infection. However, in vivo studies suggest that fewer than 1% of intestinal B lymphocytes become infected (Jones et al., 2014). These findings indicate that productive MNV infection in mice likely requires the involvement of multiple cell types (Estes et al., 2019).
Even though B lymphocytes are not the first line of defense against intestinal pathogens, the detection of viral genome in the spleens of neonatal mice infected with MNV-1 suggests that this organ (Roth et al., 2020), and possibly B cells within it, may be susceptible to infection.
Since primary cell cultures represent a more physiologically relevant model than established cell lines, as they preserve tissue-specific features, express native receptors, and better reflect natural viral tropism and replication dynamics, we investigated whether primary cultures of murine splenic B lymphocytes were permissive for a productive MNV-1 infection. We found that this cell supported MNV production, although with limited efficiency, suggesting that specific populations of B lymphocytes may be permissive to infection. However, it remains to be determined whether B cells, located in Peyer’s patches or the lamina propria, play a relevant role in MNV infection in vivo.
Intestinal B cells are anatomically close to the primary site of MNV-1 entry and remain activated due to constant stimulation to microbial products from the resident microbiota. Bacterial components, such as LPS, can interact with MNV, stabilizing the virion and protecting it from heat degradation, which, under certain conditions, may enhance infection (Budicini and Pfeiffer, 2022). In addition, macrophage tolerance to LPS has been associated with immune exhaustion, thereby increasing the susceptibility of mice to MNV-1 infection or reactivation (Makjaroen et al., 2023). Supporting this concept, a recent study reported a correlation between the abundance of LPS-rich bacteria and HuNoV infection in South African infants (Kgosana et al., 2025). Based on this evidence, B lymphocytes were stimulated with LPS and IL-4 to mimic an activated immune environment; consistent with previous reports, we observed that immune stimulation promotes MNV-1 replication.
In contrast, LPS has been reported to inhibit MNV-1 replication in macrophages, likely through inflammasome activation, highlighting a cell-type-dependent response (Yu et al., 2020). Interestingly, while the NLRP3 inflammasome can be activated in B lymphocytes by β-glucan or CpG, LPS alone appears insufficient to trigger this pathway (Ali et al., 2017). Although inflammasome signaling was not directly evaluated in the present study, a comparable model demonstrated that inflammasome activation differs between macrophages and B lymphocytes. In that context, Salmonella was shown to modulate IL-1β and IL-18 secretion as well as NLRC4 inflammasome activation in B cells, whereas LPS alone did not induce inflammasome activation; in contrast, flagellin stimulation was effective (Perez-Lopez et al., 2013). Collectively, this evidence suggests that inflammasome activation is unlikely to be a major consequence of LPS stimulation in our B-cell model under the conditions tested. Nevertheless, whether MNV-1 modulates the inflammasome pathways in B lymphocytes remains to be determined.
Conclusions
This work demonstrates that MNV-1 can infect murine splenic B lymphocytes and that LPS and IL-4 enhance viral particle production. Although the underlying mechanism remains unclear, this finding could contribute to improving MNV culture models in B lymphocytes.
Authors’ Contributions
Conceptualization: L.S.A. and A.L.G.E.; Data curation: C.E.M.R.; Formal analysis: C.E.M.R.; Funding acquisition: L.S.A. and A.L.G.E.; Investigation: C.E.M.R. and J.I.A.B.; Methodology: C.E.M.R. and J.I.A.B.; Resources: L.S.A. and A.L.G.E.; Supervision: L.S.A. and A.L.G.E.; Validation: C.E.M.R.; Visualization: C.E.M.R. and A.L.G.E.; Writing original draft: C.E.M.R.; Writing, reviewing, and editing: L.S.A. and A.L.G.E. All authors have read and approved the final version of the article. The authors declare that they have contributed significantly to the work reported, in accordance with the CRediT taxonomy detailed above.
Footnotes
Acknowledgments
We thank Clotilde Cancio Lonches and Hector Romero Ramírez for their technical assistance and Juan Ludert for his critical comments on the article.
Funding Information
This work was supported by the Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCYT), Grant no. 302965, PRONAII 3: Infecciones virales del tracto gastrointestinal, and Grant no. CF-2023-I-741. C.E.M.R. received a CONAHCYT scholarship .
Author Disclosure Statement
No competing financial interests exist.