
Abstract
It is widely known that numerous enveloped viruses can produce multinucleated cells (syncytia) as a result of viral entry-related membrane fusion events. By protecting the virus from the host’s immune reaction, these syncytia are thought to promote viral reproduction. Syncytia are collections of merged cells. A viral spike protein (S) on the surface of an infected cell interacts with receptors on nearby cells to cause the syncytia response. The innate immune system’s response to viruses affects how syncytia form. Some interferon-stimulated genes change the membrane in a way that reduces the likelihood of fusion. The severe acute respiratory syndrome coronavirus (SARS-CoV-2) virus is quickly changing; also, several mutations occurred in its S protein. Individually and together, the Alpha, Beta, Gamma, and Delta variants carry mutations that significantly affect S function and syncytia formation. The function of syncytia in newly emerging variant diseases is still unknown, though. Syncytia could cause disease through promoting viral transmission, cytopathicity, immunological evasion, and inflammatory responses. The SARS-CoV-2 S protein variations include several changes that improve receptor interactions, fusogenicity, and antibody reactivity. A wide range of clinical symptoms, including moderate febrile sickness, severe respiratory distress, and occasionally deadly lung damage, can be brought on by an infection with SARS-CoV-2. Several of these lung illnesses (MERS-CoV) are linked to both the Middle East Respiratory Syndrome (MERS) and the severe acute respiratory syndrome coronavirus (SARS-CoV). Compared to acute respiratory syndromes, the lung thrombosis brought on by Coronavirus disease 2019 (COVID-19) is incredibly severe. In this review we focused on innate immunological elements that prevent syncytia from forming and the molecular triggers of S-mediated fusion.
Background
Severe acute respiratory syndrome (SARS) coronavirus 2 is a new coronavirus that causes coronavirus disease 2019 (COVID-19) that was found in Wuhan, China (SARS-COV-2). By March 2022, it had been determined that SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) was responsible for the COVID-19 pandemic, which had caused 475 million illnesses and 6.1 million deaths (Arora, Kempf, Nehlmeier et al., 2021). Infants and young children typically contract minor upper respiratory infections from coronaviruses (CoVs) HCV-229E, HCOV-OC43, HCOV-NL63, and HKU1, but older people occasionally contract severe illnesses from these CoVs (Asarnow et al., 2021). Acute respiratory syndrome with severity, since they can infect the lower respiratory tracts and result in serious respiratory issues in people, the Middle East Respiratory Syndrome (MERS-CoV) and SARS-CoV are more deadly (Bauer, Ferla, Head et al., 2018). Like MERS and SARS, it spreads quickly (Braga, Ali, Secco et al., 2021). A highly pathogenic SARS-CoV-2 infection might result in severe flu-like symptoms that can induce acute respiratory distress syndrome, diffuse alveolar damage (DAD), pneumonia, renal failure, and even death(Buchrieser, Degrelle, Couderc et al., 2019). Fever, cough, and dyspnea were the most prevalent symptoms, seen in 83%, 82%, and 31% of 99 COVID-19 patient (Chen, 2006).
The pathogenic characteristic syncytia found in the lung tissues of patients with infections are currently being studied (Chen et al., 2020). However, an unprecedented amount of study was conducted on the molecular and viral components of coronavirus-induced syncytia formation as well as its possible effect on viral transmission, pathogenesis, and immunological responses in the past year. Syncytia is one COVID-19 feature that might be investigated. This review’s objective is to gather the most recent findings on SARS-CoV-2-induced syncytial development. It aims to characterize the components of innate immunity that prevent cell-cell fusion, offer the clinical evidence of syncytial formation by a coronavirus, and analyze the molecular variables that affect S-protein-mediated fusion. The study also looks at the virological and molecular effects of S protein mutations carried by novel fusion concerns. Finally, it discusses how virus-induced syncytial development may affect viral infection, dissemination, the immune system, and toxicity (Bussani, Schneider, Zentilin et al., 2020).
Formation of syncytia
Two or more cells fuse together to form large multinucleated cells known as syncytia. Different cells’ cytoplasmic contents and plasma membranes combine to form a single lipid bilayer during the creation of syncytia. The development of muscle fibers and the placental barrier, as well as the differentiation of bone osteoclasts, are all dependent on cells undergoing syncytialization (Rajah et al., 2022). Fusogenics are a class of specialized proteins that aid in overcoming energy and environmental barriers that prevent cellular plasma membrane fusion (like those from enveloped viruses and cellular fusogens like HAP2/GCS1) (Rajah et al., 2022). Mechanistically, cell-cell fusion is similar to virus-cell fusion. Additionally, the membranes of enveloped viruses need to fuse with cellular membranes in order for viral components to reach the cytoplasm and start the viral life cycle. The placenta’s development serves as a dramatic example of how cell-cell fusion and the mechanics of virus-cell fusion during infection are related processes. The essential placental barrier is constructed by the syncytiotrophoblast, a layer of linked cytotrophoblasts. Syncytia, the fusogenic that encourages cytotrophoblast fusion, was created by endogenous retroviruses, whose genes were inserted into mammalian genomes 10–85 million years ago (Rajah et al., 2021).
The novel coronavirus (SARS-COV-2): syncytia development
Viruses that naturally cause syncytia
Several viral families have the capacity to generate syncytia in patient-infected cells and nearby uninfected cells (Fig. 1). Regardless of the presence of other viral proteins, a viral fusion protein (commonly termed F) promotes this process during infection. To date, researchers have focused on studying naturally occurring fusogenic viruses such as the measles, Sendai virus, respiratory syncytial virus, and Newcastle disease virus. In addition to these viruses, some other viral families, such as retroviruses, can cause viral particles and cellular membranes to fuse without resulting in the development of syncytia (Burton and Bartee, 2019).
Syncytia are formed by the fusion of infected cells with neighboring cells, leading to the formation of multi-nucleate enlarged cells. This event is induced by the surface expression of a viral fusion protein that is fusogenic directly at the host cell membrane.
There is evidence that coronaviruses can cause syncytia
Acute respiratory distress, as well as severe, occasionally fatal lung damage, are all possible clinical indications of SARS-CoV-2 infection. Alveolar injury, micro/macrovascular thrombosis, and pneumocyte necrosis are the three primary lung diseases. These illnesses resemble those brought on by infections with the CoVs that cause SARS and the MERS-CoV. When compared to other conditions that result in acute respiratory syndrome, particularly when considering the widespread lung thrombosis, COVID-19’s clinical symptoms are highly severe. The epithelial lining of the lungs deteriorates due to hypoxemia, immune-mediated damage, and viral-induced cytopathy (Bussani et al., 2020; Guzik et al., 2020; Braga et al., 2021). An immunological reaction probably causes the majority of the documented histopathology (Bussani et al., 2020). An infection of pulmonary epithelial cells can trigger the activation of monocytes, macrophages, and dendritic cells, along with the production of proinflammatory cytokines. Blood from those with the most severe types of the condition contains an excessive amount of cytokines. Virus-induced cytopathies can cause direct lung tissue damage as well as immune system activation. Also noticed in numerous clinical cases are multinucleated pneumocytes (Bussani et al., 2020).
In cell culture models, many “common cold” CoVs, such as hCoV-HKU1, hCoV-NL63, and hCoV-229E, cause syncytia. To the best of our knowledge, these widely dispersed CoVs have not been linked to any reported cases of syncytia. There are fewer autopsy reports because they are not thought to be fatal. Therefore, testing the syncytia of CoVs that cause the common cold in animal models is necessary. Whether or how pathophysiology is impacted by SARS-CoV-2-mediated syncytia development is yet unknown. It might, however, aggravate the deleterious effects of virus-induced immune-mediated and cytopathic damage. In extreme circumstances, it causes lung tissue to deteriorate. Additionally, syncytia may facilitate the transmission of the virus and immune system evasion by protecting the virus from immune cells and neutralizing antibodies (Kanai et al., 2019; Rajah et al., 2022). In contrast, the relevant effects of SARS-CoV-2 include coagulopathy and syncytial development. Nevertheless, the fundamental chemical processes that activate these forms are still not fully understood. The S glycoprotein in the cytoplasmic space has been found to contain a proven design in this instance. The 561,000 proteins, 79 contained this verified design (UniProt bank). The SARS-CoV-2 genes NSP1 and ORF6 proteins initiate COVID-19’s pathophysiology and set off a devastating cascade of errors (Matsuyama et al., 2021). These viral ingredients cause Signal Transducer and Activator of Transcription 1 (STAT1) dysfunction, while STAT3 is hyperactivated as a defense mechanism. Infected cells with SARS-CoV-2 may experience an escalating cycle of activation due to a positive feedback loop between STAT3 and plasminogen activator inhibitor-1 (PAI-1). This is comparable to the interdependent signaling networks affected by COVID-19. Especially, coagulopathy with intravascular thrombi is caused by overexpressing PAI-1(Matsuyama et al., 2021). When too much PAI-1 binds to TLR4 on macrophages, pro-inflammatory cytokines and chemokines are released. The recruitment and subsequent activation of innate immune cells within an infected lung leads to a hypoxic environment that further encourages PAI-1 production, resulting in the infection of localized endothelial cells. Acute lung injury also phosphorylates STAT3 and activates estimated glomerular filtration rate (EGFR). Autopsies of COVID-19 patients frequently show increased production of hyaluronan and DAD, along with higher levels of PAI-1. COVID-19 risk factors are consistent with this circumstance given that PAI-1 levels are elevated in conditions such as hypertension, obesity, diabetes, cardiovascular diseases, and old age (Matsuyama et al., 2021).
The S protein mediates fusion through its molecular mechanism and associated proteins
S protein has 1271 amino acids and functions as a viral protein (Fig. 2). The S1 subunit and the S2 subunit, two functional components of the S protein, are in charge of binding to the host cell receptor. S is often split at the boundary between S1 and S2 in many CoVs, although it still has non-covalent bonds while it is in the prefusion state (Chan et al., 2013; Buchrieser et al., 2019).
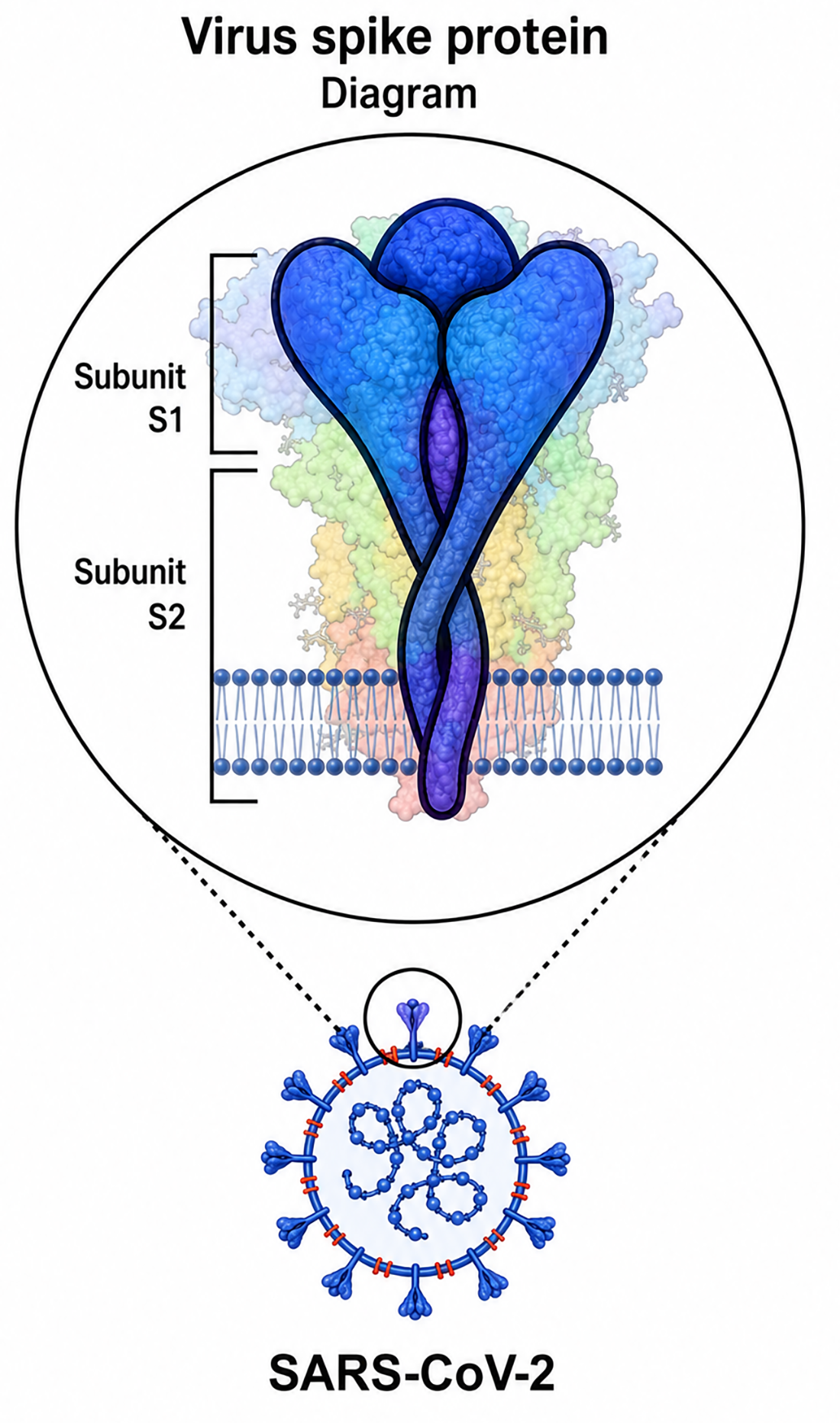
Some coronaviruses cleave their S protein into S1 and S2 subunits during their biosynthesis in infected cells, but other coronaviruses cleave their S protein only when they reach the cell. MERS-CoV, like SARS-CoV-2, belongs to the first category. Its S protein is cleaved by protein convertases such as furin in virus-producing cells. Thus, the S protein on the mature virion is composed of two non-covalently bound subunits; the S1 subunit that binds to ACE2 and the S2 subunit that binds to the membrane. Furthermore, the S2 subunit contains a fusion peptide and other machinery needed for membrane fusion upon infection.
Areas where variants of concern have been documented (as of August 31, 2021). Nine colored patches indicate the presence of the Alpha, Beta, Gamma, and Delta variations.
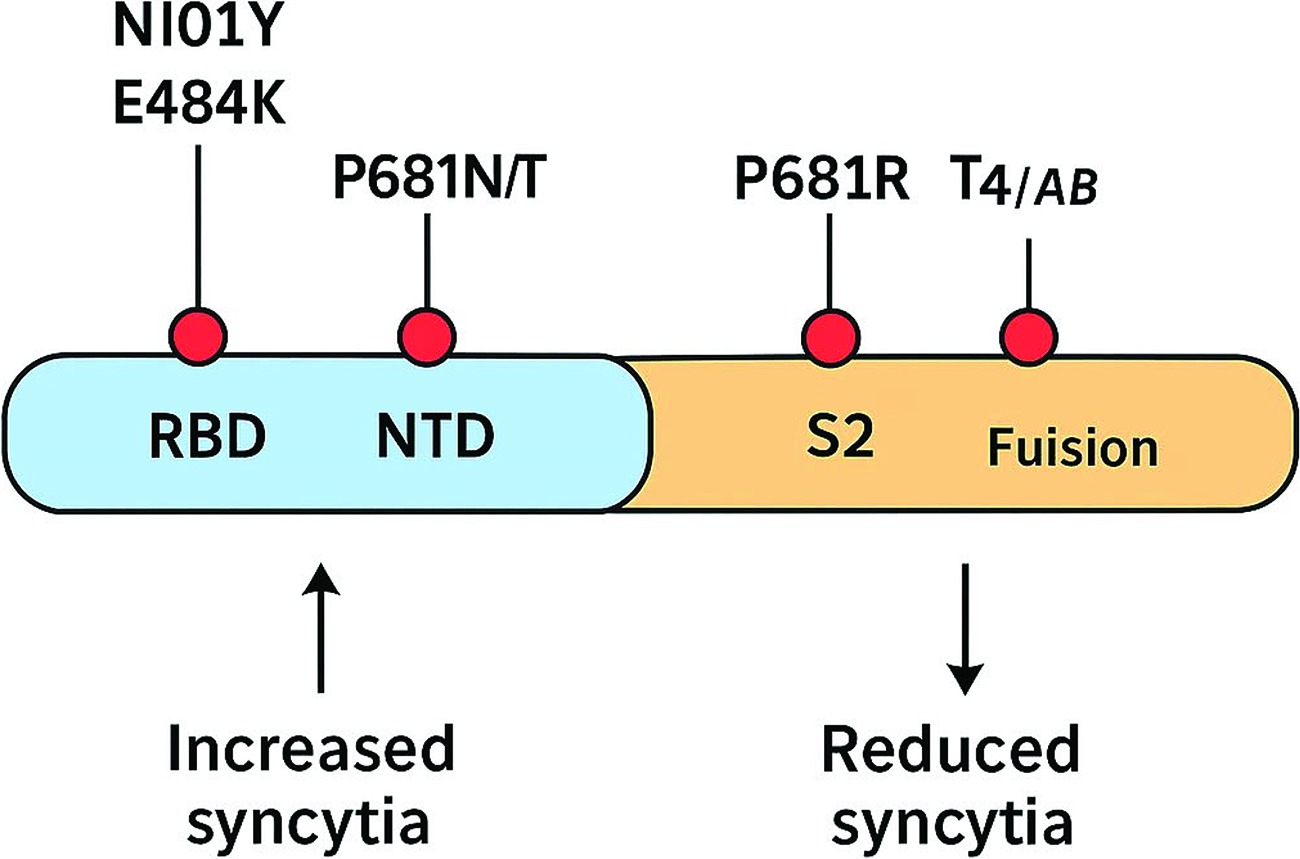
Schematic representation of major spike protein mutations influencing syncytia formation across SARS-CoV-2 variants.
Summary of Major SARS-CoV-2 Spike (S) protein Mutations and Their Reported Effects on Syncytia Formation
Soon after the SARS-CoV-2 Omicron lineage BA.1 was created and disseminated over the world, BA.2, another Omicron lineage, began to outcompete BA.1. The results of investigations showed that BA.2’s viable generation number was 1.4 times greater than BA.1’s. Moreover, neutralization testing showed that resistance sparked by widely administered COVID vaccines against human populations was ineffective against BA.2. BA.2’s antigenicity was noticeably different from BA.1’s. Cell culture experiments showed that the BA.2 spike was superior to the BA.1 spike in interceding syncytia organization and conferred greater replication viability in human nasal epithelial cells (Yamasoba et al., 2022). Hamster illness experiments indicated that the BA.2 spike-bearing infection was more pathogenic than the BA.1 spike-bearing infection. Three primary lineages have been identified in Omicron: BA.1, BA.2, and BA.3. The S protein of BA1.1, a sublineage of BA.1, has an R346K alteration. BA.1 differs from BA.2 by 50 amino acids, approximately twice as many as the four other variations of concern (Alpha, Beta, Gamma, and Delta) and Wuhan-Hu-1, a representative SARS-CoV-2 disconnect (Yamasoba et al., 2022).
RBDs are found in the distal S1 subunits. They aid in maintaining the fusion machinery-containing S2 subunits in a prefusion state (Gui et al., 2017). Just upstream of the fusion peptide (FP), at a location known as S20, host proteases further cleave all CoVs (Madu et al., 2009).
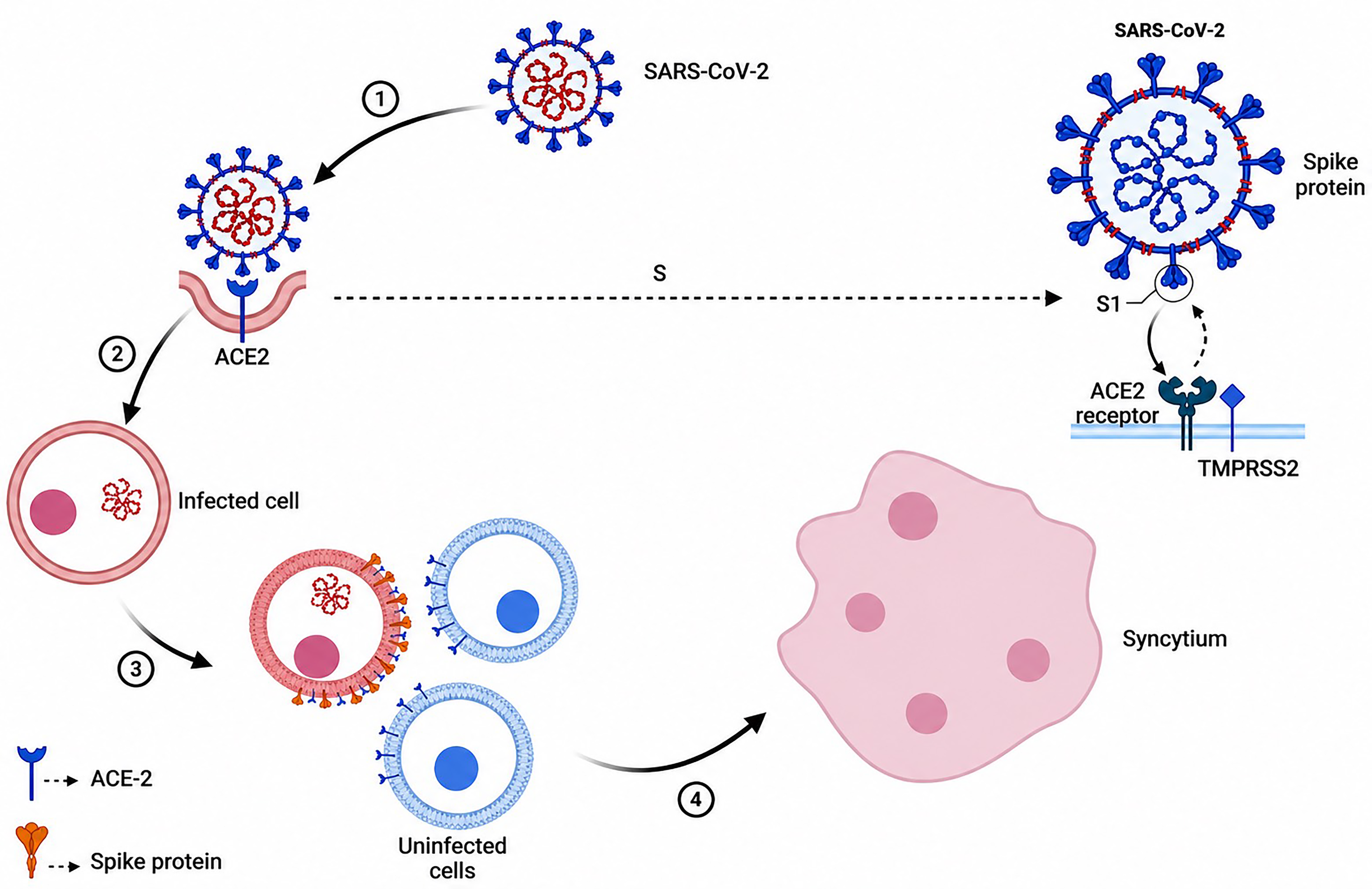
It has been hypothesized that the syncytial arrangement of pneumocytes with multinucleation, which is a necrotic hallmark in the lungs of COVID-19 patients, results from the interaction of cells transmitting the S protein and its ACE2 receptor (Jocher et al., 2022). Since S and ACE2 are both necessary for lung cell contamination, it is possible that ADAM10 and ADAM17 are required for syncytia formation and that metalloprotease inhibitors can prevent this from happening. A549-ACE2 NTC cells were co-cultured with HEK293T cells that were expressing both S and GFP to test this theory, and the results showed massive GFP-positive syncytia. The GFP-positive syncytia zone was impressively reduced by more than 50% of the cores present in each syncytium as a result of the expansion of Batimastat (BB94). In A549-ACE2 cells, a single knockout of ADAM10 was sufficient to reduce the number of cores in syncytia and the arrangement of syncytia to that of NTC cells that had received BB94 treatment. The single knockout of ADAM17 during differentiation had no effect on syncytia arrangement, and the double knockout of ADAM10 and ADAM17 had an effect similar to that of the single knockout of ADAM10. Furthermore, the lysosomal inhibitor E64d, which successfully prevented SARS-CoV-2 cell contamination, did not disrupt cell fusion. This suggests that syncytial arrangement depends on ADAM10 and does not require lysosomal proteases in differentiation to lung cell disease, which requires ADAM17 and lysosomal proteases (Jocher et al., 2022).
As a result, different sets of proteins regulate syncytia organization and cell contamination throughout COVID-19 pathology. The SARS-CoV-2 S protein is taken into consideration because both forms are encouraged following the proteolytic preparation of S, even though both steps necessitate the expression of ACE2 and S to determine the protease substrate that may be pertinent for ADAM17-dependent SARS-CoV-2 cell fusion and ADAM10-dependent lung cell fusion. By the cleavage of S at a position known as the S2 site inside the S2 subunit, TMPRSS2 may enhance SARS-CoV-2 infection. Although SARS-CoV-2-infected cells like A549-ACE2 do not express TMPRSS2, as seen in single-cell RNA-seq data, other cryptic proteases are also likely to contribute. Moreover, the growth of exogenous proteases like trypsin or TMPRSS2 improves syncytia organization, likely through S protein cleavage (Jocher et al., 2022). The question of whether recombinant ADAM10 and ADAM17 can prime the S protein similarly to TMPRSS2 remains open. As ADAM10 or ADAM17 increased, results showed an atomic weight shift from 70 to 55 kDa, consistent with what was observed near TMPRSS2. The discovery of several S2’ cleavage sites with relatively low molecular weight for ADAM10 suggests multiple cleavage sites near the known S2’ locus. In a cell-free experiment, ADAM10 and ADAM17 together can cleave the S protein similarly to TMPRSS2 (Jocher et al., 2022).
Cell-to-cell fusion plays a role in SARS-CoV-2 syncytia production because optimization of cleavage sites S1/S2 encourages cell-to-cell fusion but not virus-to-cell fusion. Viral entry is still possible even if the S1/S2 furin cleavage site is removed (Chen, 2006). Infection with SARS-CoV-2 has been linked to the S1/S2 furin cleavage site, particularly during syncytia formation. The RBD and N-terminal domain (NTD) comprise the S1 subunit. While the NTD’s role is not fully understood, studies on other CoVs suggest it may be involved in glycan recognition during attachment, receptor recognition, and pre-to-post fusion transition of the S protein. DC-SIGN and L-SIGN lectins may also interact with NTD as SARS-CoV-2 receptors. Antibodies can facilitate S protein-mediated fusion by inducing an open RBD state upon binding to NTD (Madu et al., 2009).
The S protein trimer’s “up” (open) and “down” (closed) RBD conformations control cellular tropism and interact with ACE2 receptors. ACE2 binds to the “up” RBD, sequentially opening remaining RBD monomers for binding. This puts the S protein under mechanical stress (Madu et al., 2009). ACE2-bound S1 monomers separate during opening, exposing the trimeric S2 core housing the fusion machinery. The S2 domain contains the FP, heptapeptide repeats (HR1 and HR2), transmembrane anchor (TA), and C-terminal domain (CTD). HR1 forms a helix after S1 release and FP insertion into the target membrane, disrupting the lipid bilayer to connect target and fusion membranes. HR1 and HR2 interact to form a six-helix bundle, overcoming natural repulsion, producing hemi-fusion, and enabling cytoplasmic mixing. CTD is linked to intracellular trafficking via COPI/II, and syncytia generation depends on the cysteine-rich cytoplasmic region of S protein (Madu et al., 2009).
S protein subdomains are essential for receptor recognition, binding, translocation, and fusogenicity. During the pandemic, S protein evolution resulted in multiple variants of concern (VOCs) with mutations affecting these functions (Zhang et al., 2007). SARS-CoV-2 S protein structure and function remain under investigation (Riccio et al., 2022). Full-length SARS, MERS, and SARS-CoV-2 S plasmids, either untagged or with a C-terminal tag, were used to characterize S protein biogenesis in HEK293T and A549 cells. Proteolytic processing at the S1/S2 cleavage site was confirmed using the furin inhibitor RVKR, showing efficient S protein cleavage in human lung cells (Riccio et al., 2022).
HeLa-ACE2 cells transfected with SARS-CoV-2 S protein formed multinucleate syncytia, enhancing cell-to-cell fusion (Ren et al., 2021). At 24 h post-transfection, syncytium formation reached ∼60%, with transfected cells showing micronuclei in >23% of all cells and >93% of syncytia. The average syncytium contained four micronuclei, correlating syncytium size with micronuclei number. Similar S1 CTD/NTD receptor-binding arrangements were observed in other CoVs, including MERS-CoV and SARS-CoV, whereas mouse hepatitis CoV uses S1 NTD (Li et al., 2005; Lu et al., 2013).
Infected cells merge with neighboring cells as the SARS-CoV-2 infection progresses and viral S protein is produced at the cell surface, rupturing the nuclear membrane and leading to the formation of micronuclei. A micronucleus is recognized by the second messenger cyclic GMP-AMP (cGAS), which then activates the cGAS receptor stimulator of interferon genes and phosphorylates interferon regulatory factor 3 to produce interferons. This mechanism should work in conjunction with others, including TLR3, RIG-I, and melanoma differentiation-associated protein 5, which are pattern recognition receptors that recognize viral RNA and produce IFNs, inflammatory cytokines, and chemokines. Syncytia are produced as a result of the host proteases’ proteolytic activation of the viral S protein. The proteolytic activation of the S protein takes place in two distinct places (Buchrieser et al., 2020; Koch et al., 2021; Mykytyn et al., 2021). In a single activation step, the S protein is cleaved at the S1–S2 border. At the S2′ location, a second proteolytic activation takes place. Due to their potential to increase the infectiousness of cell-free virions and the expulsion of lymphocytes by multinucleated pneumocytes, host proteases that cleave the SARS-CoV-2 S protein may play a role in COVID-19’s pathophysiology (Zhang, Zheng et al., 2021). These results suggest that host protease facilitates S protein cleavage to modulate innate immune activation. Given the significance of syncytia in the pathophysiology of COVID-19, neutralizing antibodies’ apparent impairment of syncytium development (Asarnow et al., 2021), drugs (Braga, Ali et al., 2021), or protease inhibitors (Cheng et al., 2020) may lead to increased effectiveness against advanced or serious illnesses (Liu, Wei et al., 2022).
Syncytia formation during infection with SARS-CoV-2 variants of concern
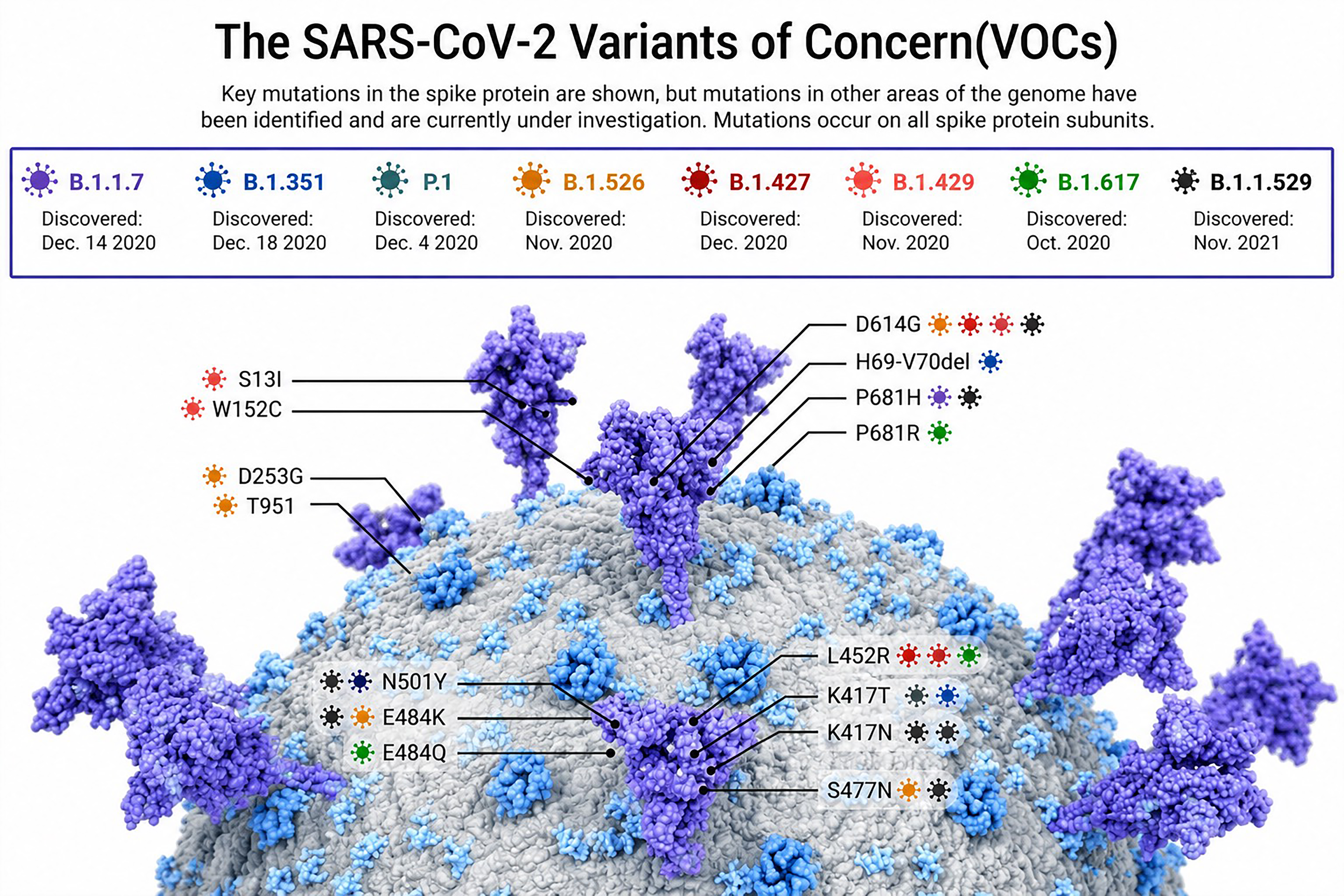
Since the start of the pandemic, mutants with multiple mutations throughout the genome have displaced the original Wuhan strain (Korber et al., 2020; Davies et al., 2021). Brazilian Gamma (P1 and P2), Indian Delta (B.1.617.2), South African Beta (B.1.351), and British Alpha (B.1.1.7) are important regionally and internationally. All throughout the viral genome, there are mutations connected to these variants, many of which involve the S protein.
European mutants with the D614G mutation near the S1/S2 cleavage region were among the first discoveries. Although this mutation modifies the S protein’s structure to make it more ACE2 binding-competent, it does not impact antibody neutralization (Yurkovetskiy et al., 2020; Zhou et al., 2021). In primary cell culture models, the D614G variant replicates more efficiently, and upper respiratory tracts of patients frequently contain D614G, which may be linked to infection rather than disease severity (Davies et al., 2021).
Pseudovirus assays for fusogenicity revealed that D614G boosts cell penetration efficiency and generates more syncytia than the original Wuhan strain. Alpha, Beta, and Delta variants also produce more syncytia, with Alpha and Delta S proteins inducing the majority of cell-cell fusions faster than other variants. The Beta variant S protein is more fusogenic than D614G. Syncytia formation appears strongly correlated with the variant S proteins’ ACE2 affinity (Saito et al., 2021). Some studies suggested that variant S proteins cause cell-cell fusion with little or no change (Alpha, Beta, and Gamma), while Delta was more syncytiogenic (Arora et al., 2021; Hoffmann et al., 2021). Experimental setup and timing differences may explain this disparity. D614G alone significantly alters S protein structure, receptor affinity, and fusogenicity, highlighting the importance of evaluating other variant-associated mutations (Arora et al., 2021).
The S1 domain of the spike is the main target for neutralizing antibodies and is under constant evolutionary pressure. Alpha, Beta, and Gamma variants include the RBD mutation N501Y, which increases ACE2 affinity and viral transmission without impairing neutralization (Yurkovetskiy et al., 2020; Saito et al., 2021; Zhou et al., 2021). The N501Y mutation strengthens interaction with the ACE2 hydrophobic pocket (Y41 and K353). Although N501Y alone does not enhance syncytia formation, the Alpha lineage partially carries the E484K mutation, found in Beta and Gamma, along with K417N in Beta (Collier et al., 2021). N501Y, E484K, and K417N may affect ACE2 binding and facilitate antibody escape, with E484K alone enabling infection of H522 human lung cells independently of ACE2 (Puray-Chavez et al., 2021).
Variant S proteins also carry multiple NTD mutations linked to antigenicity and fusogenicity (Puray-Chavez et al., 2021). The Alpha 69/70 deletion increases virion infectivity and compensates for reduced RBD activity due to antibody escape (Meng et al., 2021). Loss of 69/70 in D614G negatively impacts syncytia formation (Davies et al., 2021). The Beta NTD deletion 242–244 reduces syncytia formation (Davies et al., 2021), while D215G enhances it. These mutations may influence binding to L-SIGN, DC-SIGN, neuropilin-1, and overall S structure (Soh et al., 2020).
The P681H (Alpha) and P681R (Delta) mutations at the S1/S2 cleavage sites enhance furin cleavage and syncytia formation (Soh et al., 2020; Saito et al., 2021). P681R in Delta increases cleavage and viral fusion, enhancing pathogenicity in hamster models (Saito et al., 2021). The Alpha S2 D1118H mutation also promotes syncytia, though mechanistic details are limited (Saito et al., 2021).
Omicron infection may favor endosomal fusion via cathepsins due to low syncytia formation. Omicron S protein exhibits reduced fusogenicity in vitro, with pseudotyped virus showing minimal syncytia in TMPRSS2-expressing cells compared to delta or WT/D614G (Zhang et al., 2022). Despite high ACE2 binding and effective S1/S2 cleavage, S2 mutations delay fusion, and endosomal entry increases susceptibility to restriction factors, possibly contributing to milder lung cell infection (Zhang et al., 2022).
Membrane cholesterol is required for SARS-CoV-2-infected cells
Cell fusion studies of SARS-CoV-2 infection using U2OS and VeroE6 cell lines have shown that lowering cholesterol in the plasma membrane using various effective compounds prevents cell fusion (Hoffmann et al., 2020). These compounds are predicted to be ineffective in virus entry models but are necessary to disrupt viral membranes. Methyl-beta-cyclodextrin (MBCD), frequently used to deplete cells of cholesterol, blocks viral entry by disrupting plasma membrane cholesterol without targeting intracellular mechanisms. This effect is observed more strongly in pseudo-virus entry models than in syncytia assays. By lowering cholesterol in the viral membrane (not the host cell), viral entry can be inhibited (He et al., 2018; Hu et al., 2019). Without treatment, viral infection produces multinucleated cells, whereas low micromolar doses of MBCD prevent infection. Therefore, the cholesterol content of SARS-CoV-2 particles is crucial for infectivity (He et al., 2018).
Fusion of the spike is mediated by membrane cholesterol through a raft-independent mechanism
For membrane fusion to occur, spike interaction with specific plasma membrane components is necessary. Therefore, such assemblies have slower dynamics compared to transmembrane proteins, which diffuse more freely in a two-dimensional lipid bilayer (Bauer et al., 2018). An intriguing hypothesis is that membrane-proximal spike regions control diffusivity and fusogenic behavior by promoting the interaction of membrane domains rich in cholesterol, sometimes called “lipid rafts” (Simons and Ikonen, 1997; Pelkmans and Helenius, 2003; Levental and Levental, 2015). Because palmitoylation of proteins may facilitate their association with 10–50 nm protein–lipid complexes in the plasma membrane, spike cysteine residues are especially crucial (Levental et al., 2010). The SARS-CoV-2 spike partitioned less firmly into the dense, organized phase of giant plasma membrane vesicles (GPMVs) in earlier trials. The phase contains large amounts of sphingolipids and cholesterol (Levental et al., 2009, 2011). They cannot rule out that living cells have different lipid raft properties from GPMVs, but they suggest that the S protein participates in membrane fusion and may depend on the palmitoylation of its cysteine-rich CTD (Levental et al., 2020). Numerous studies have shown that cholesterol-rich membrane elements adhere well to glass surfaces and are often sloughed off from cells (He et al., 2018; Hu et al., 2019).
**Host factors affecting syncytia formation**
Various host factors complement the fusogenic capability of the S protein, influencing syncytia formation. Interferon-induced transmembrane (IFITM) proteins are small transmembrane proteins expressed by interferon-stimulated genes (ISGs) and can block viral infections by preventing viral membrane fusion. Reports suggest that IFITMs may diminish SARS-CoV-2 infection; however, conflicting findings exist. Expression of IFITMs effectively inhibited SARS-CoV-2-induced syncytia, with IFITM1 exhibiting the greatest potency (Bauer et al., 2018). The expression of the serine protease TMPRSS2, which facilitates S-induced syncytia, may counteract this inhibitory effect. In vitro models indicated that both IFITMs and TMPRSS2 affect fusion efficiency when co-localized with ACE2 within the same cell rather than in the S-expressing cell (Bauer et al., 2018).
Tri- and tetra-thioredoxin mimetic (TXM) peptides obstruct syncytia formation and the interaction of the SARS-CoV-2 spike protein with the ACE2 receptor. The RBD of the S1 subunit contains four pairs of cysteine residues, which are influenced by the redox state of the extracellular environment, thereby affecting the spike protein’s affinity for ACE2 (Govednik et al., 2024). Researchers evaluated thiol-based drugs, including N-acetylcysteine amide (AD4/NACA) and TXM peptides, aiming to reduce disulfide bonds in the RBD. AD4/NACA is an amide derivative of N-acetylcysteine (NAC) with greater efficacy and membrane permeability than NAC. This research was also contributed to by Atlas (2021), Bartov et al. (2006), Grinberg et al. (2005), and Offen et al. (2004). TXM peptides are low molecular weight thiol-reducing peptides, designed according to the -Cys-X-X-Cys- and -Cys-X-Cys motifs that enhance the redox activity of Trx1 and other oxidoreductases (Govednik et al., 2024). The X in these motifs may denote any amino acid residue. The presence of two cysteines in tri- and tetra-TXM peptides promotes reduction of protein disulfides, mimicking Trx1 function (Govednik et al., 2024).
Conclusion
Histological studies revealed that the lung tissues of COVID-19 patients contained infected multinucleated syncytial cells. The influence of syncytia on disease is incompletely comprehended. Recent studies have elucidated the role of syncytia in SARS-CoV-2 infection and the mechanisms behind their creation. Viruses can spread from cell to cell or after a cell dies. They can also target immune cells and evade antibodies that would normally kill them by hiding. Rapid syncytial collapse, on the other hand, can prevent viral multiplication and trigger an inflammatory immune response. It is highly likely that the SARS-CoV-2 syncytia will increase viral pathogenicity. Syncytia are vulnerable to the antiviral response of the innate immune system. Sometimes ISGs can change the membrane to stop fusion. The SARS-CoV-2 virus’ S protein is rapidly evolving and has undergone multiple changes. S function and syncytia formation are significantly affected by mutations discovered in the Alpha, Beta, Gamma, and Delta variants, both individually and collectively. About how syncytia influences the pathophysiology of emerging variants, there is still much to discover.
Authors’ Contributions
A.T. collected the related articles and drafted the article. R.D. and R.J. revised the article. R.D. and R.J. participated in the design of the article. All authors have read and approved the final article.
Footnotes
Acknowledgments
The authors would like to thank Ms. A. Keivanshekouh at the Research Consultation Center (RCC) of Shiraz University of Medical Sciences for her invaluable assistance in editing the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding were received for this article.