
Abstract
Pelizaeus-Merzbacher disease (PMD) is an X-linked recessive genetic disorder. Patients with PMD usually present with dystonia, ataxia, progressive spasms, nystagmus, and motor developmental delay, which may be misdiagnosed as cerebral dysplasia. We summarize the clinical and genetic characteristics of 4 patients to explore the early diagnosis of PMD. Three male patients from one family and 1 male patient from another family were included. Cases 1 to 3 presented with intellectual disability, motor delays, ataxia, and nystagmus. They were diagnosed with classical PMD via whole-exome and copy number variation (CNV) testing. Case 4, a 1-year-old with similar symptoms, carried a c.737G>A mutation in PLP1 but showed no CNVs. All patients underwent rehabilitation. PMD can be diagnosed early by chromosomal CNV detection. Classic PMD is the most common form of PMD, with PLP1 gene duplication being the leading cause. Although rare, point mutations in PLP1 can result in classic PMD.
Pelizaeus-Merzbacher disease (PMD) is a rare type of progressive X-linked recessive leukodystrophy characterized by dysmyelination in the central nervous system (CNS). The disease primarily affects males and results in a spectrum of neurologic deficits, including nystagmus, hypotonia, ataxia, spasticity, and variable degrees of intellectual disability. Morlet et al 1 suggested that PMD is caused by different types of mutations in proteolipid protein 1 (PLP1), specifically point mutations, gene duplications, and gene deletions. The PLP1 gene encodes 2 major membrane proteins, PLP1 and its smaller isoform DM20, which are the most abundant constituents of myelin in the CNS. Consequently, dysfunction of PLP1 disrupts oligodendrocyte maturation and myelin integrity, potentially leading to severe neurologic impairment. Despite recent advances in molecular genetics, the early diagnosis of PMD remains a significant clinical challenge. Its initial symptoms, such as developmental delay and nystagmus, are nonspecific and often overlap with other neurologic conditions, including cerebral palsy and nonspecific cerebral dysplasia, leading to frequent misdiagnoses and diagnostic odysseys for families. Herein, we present the cases of 3 boys from one family and that of one boy from another family, all with classic PMD, illustrating 2 distinct genetic etiologies: familial gene duplication and a de novo missense mutation. By comparing the clinical and genetic characteristics of these patients, we aim to highlight the importance of comprehensive genetic testing strategies, including copy number variation (CNV) analysis and whole exome sequencing (WES), in achieving early and accurate diagnosis. This study was approved by the Ethics Committee of the Seventh Affiliated Hospital of Sun Yat-sen University (No. KY-2023-025-01). The father of the 3 boys and the parents of the other boy provided informed consent to the research and agreed to the publication of the results.
Patients and Methods
General Information
In August 2020, the 3 boys visited the outpatient clinics of the Pediatric Department of Fengqing People's Hospital of Yunnan. Case 1 was the elder brother, whereas cases 2 and 3 were twins and the younger brothers of case 1.
Case 4 was admitted to the pediatric outpatient clinics of the Seventh Affiliated Hospital of Sun Yat-sen University in May 2025.
Case 1
A 6-year-old boy presented at the first visit with a 5-year history of intellectual disability and growth delays. The child was born to a G1P1 mother. The patient was delivered prematurely, but the exact gestational age and birth weight were unknown. At the age of 6 months, he developed delayed motor development, but no medical interventions were conducted. At the age of 15 months, he was admitted to Kunming Children's Hospital of Yunnan and diagnosed with intellectual disability and moderate malnutrition. Sialic acid and gangliosides were administered but were not effective. The patient was transferred to the rehabilitation center. Physical examination revealed no facial abnormalities, no particular smell, no relevant abnormalities on cardiopulmonary auscultation, small scrotums with bilaterally descended testicles, and flat feet. The patient had intermittent bilateral horizontal nystagmus. He could sit but could not walk independently. He experienced ataxia when walking with support. The Babinski sign was bilaterally positive, whereas cervical resistance, the Brudzinski sign, and the Kernig sign were negative. The walking gait is shown in Video 1. The child could understand instructions and communicate using simple words instead of short sentences.
Cases 2 and 3
The 4.5-year-old twin boys presented with a 3-year history of intellectual disability and growth delay at the first visit. The twin boys were born to a G2P2 mother. They were born prematurely, but their exact gestational age was unknown. They are the younger brother of case 1. When the twin boys were aged 1 year, they had the same symptoms as those of case 1. The patients received rehabilitation treatment when they were 2 years old. On physical examination, the twin boys developed nearly the same signs as case 1, including intermittent bilateral horizontal nystagmus (Video 2), flat feet, and bilateral positive Babinski sign. They could sit independently but could not walk. They could understand instructions and communicate using simple words instead of short sentences.
The father was aged 30 years and healthy. Referring to the father's description, the mother was aged 30 years and healthy but could not be contacted. All 3 children were supported by their healthy paternal grandparents. No family history of genetic diseases was found.
Case 4
A 1-year-old boy presented for evaluation of developmental delay, which had been noted for approximately 1 year. The delay was first observed at the age of 3 months. Subsequent genetic testing revealed a mutation in the PLP1 gene, confirming a diagnosis of PMD with an X-linked recessive inheritance pattern. The boy had a history of slow weight gain. He had previously exhibited nystagmus, which had improved by the time of presentation. On physical examination, the child had severe growth failure, with a weight of 6.7 kg (<first percentile), a length of 67.2 cm (<first percentile), and a head circumference of 43.8 cm (fourth percentile). He had a dull expression and did not track objects or faces. The profound motor delay, with an inability to achieve head control or sit, is consistent with the significant axial hypotonia observed in early PMD. This progressed to include spastic extension in both feet. Nystagmus was absent on examination. The cardiopulmonary and abdominal examinations were unremarkable, and the anterior fontanelle was closed. The child did not follow commands but could be coaxed to smile.
Laboratory and Imaging Inspections
Case 1 was admitted to Kunming Children's Hospital of Yunnan at the age of 15 months. No abnormal findings were noted in the complete blood count (CBC), stool examination, blood electrolytes, liver function, renal function, blood ammonia level, ceruloplasmin level, immunoglobulin level, T-cell subsets, complement, and TORCH antibodies. Tests for hepatitis B virus (HBV), hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Treponema pallidum were negative. Urinary genetic metabolic disease analysis revealed no abnormalities. Electrocardiography revealed sinus tachycardia. Brain magnetic resonance imaging (MRI) demonstrated delayed myelination in the brain, as shown in Figure 1.
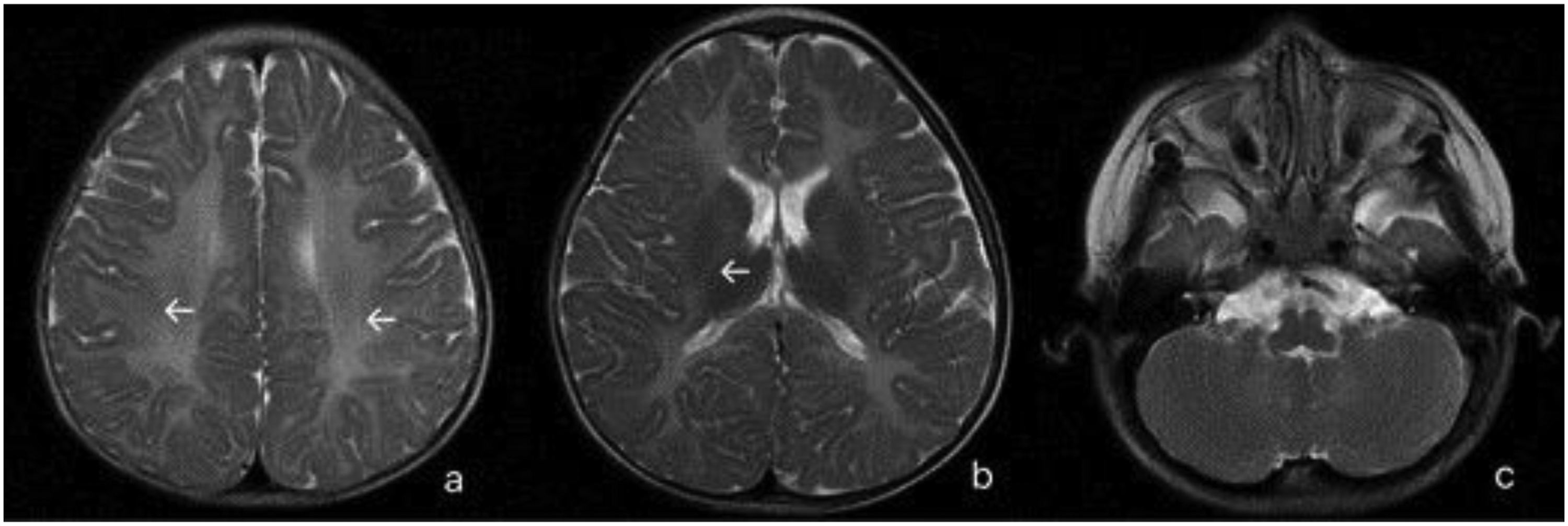
Brain magnetic resonance imaging of case 1. White arrows indicate bilateral dystrophy of the white matter in the brain.
Electroencephalography (EEG) showed increased theta activity, characterized by a frequency of 4.5-6.5 Hz and an amplitude of 20-80 μV, in the background during the waking period and no epileptiform discharge. The language development of case 1 was equivalent to that of a 5-month-old child, and gross motor, fine motor, and cognitive levels were lower than those of children of the same age.
Cases 2 and 3 did not undergo laboratory examination because of financial constraints.
The results of the laboratory examinations for case 4 showed normal CBC, electrolytes, liver function, renal function, blood ammonia, and lactic acid levels. Neonatal screening tests for congenital genetic metabolic disorders were all normal. Brain MRI showed no abnormalities, and an EEG showed no epileptiform discharges.
Results
Diagnosis
The congenital genetic and metabolic diseases, Wilson disease, TORCH infections, congenital immune deficiency, glycogen accumulation, and mucopolysaccharide diseases could be ruled out based on the blood and urine tests. Brain MRI revealed delayed myelination in the brain. The results of whole-exome sequencing (WES) using chip-capture high-throughput sequencing were as follows:
Case 1: No clinically significant variation consistent with a phenotype-related pathogenic or suspected pathogenic variation or genetic pattern was detected in the primary and secondary test results. High-throughput total external copy number variation (CNV) testing revealed the following: 46, XN, dup (Xq22.1-q22.2). seq [GRCh37/HG19] (102564473-103218540) * 2,xq22.1-q22.2 segment detected with approximately 0.65 Mb of repetition. This abnormality suggested PMD caused by the abnormal function of the OMIM gene PLP1 and X-linked spastic paraplegia type 2 (SPG2). Case 2: No clinically significant variation consistent with a phenotype-related pathogenic or suspected pathogenic variation or genetic pattern was detected in the primary and secondary test results. High-throughput total external CNV testing revealed the following: 46, XN, dup (Xq22.1-q22.2). seq [GRCh37/HG19] (102530041-103218540) * 2,xq22.1-q22.2 segment detected with approximately 0.69 Mb of repetition. This abnormality suggested that PMD was caused by the abnormal function of the OMIM genes PLP1 and SPG2.
The results of case 3 were identical to those of case 2.
Genetic testing of the father revealed no chromosomal abnormalities in the children. However, the mother's test was not performed because of a loss of contact.
Case 4: WES revealed a likely pathogenic variant in the PLP1 gene: NM_001128834.1:c.737G>A(p. Gly246Glu), which has been previously reported. 2 This variant was identified as hemizygous in the patient and was absent in his father, mother, brother, and 2 sisters, indicating a de novo mutation. This abnormality is associated with PMD (OMIM: 312080). However, no large-scale deletions or duplications that could explain the patient's phenotype were detected by high-throughput CNV testing. Genetic tests performed on his father, mother, and two sisters showed no abnormalities.
Treatment and Follow-up Outcomes
The three boys (cases 1-3) from the same family underwent rehabilitation treatment. The ophthalmologic consultation showed no abnormalities in the eyes of the children. Their language development, motor development, and cognitive level did not improve substantially. Case 4 also underwent rehabilitation treatment; similar to the other 3 cases. His motor, cognitive, and language development did not improve.
Discussion
PMD can be classified into classic, connatal, transitional, SPG2, and PLP-null types. Classic PMD is the most common type of PMD and usually occurs within 1-5 years of age, and the patients present with nystagmus, hypotonia, and ataxia, accompanied by cognitive dysfunction. Most patients with classic PMD die between the ages of 30 and 70 years. Connatal PMD is the most severe type, and transitional PMD is the intermediate type. In this study, 3 boys of the same family and another boy were diagnosed with classical PMD. Duplication of the PLP1 gene generally results in classic PMD. Meanwhile, missense mutation often leads to connatal PMD and may also cause other phenotypes, including classic PMD. 1 Another 5-level classification system (forms 0 to 4) for the clinical phenotypes of PMD was developed by Cailloux and colleagues, which is based on the highest level of motor function a patient achieves between the ages of 1 and 10 years. 3 These clinical classifications, which span a wide spectrum of severity, are ultimately rooted in the distinct molecular etiologies of the disease. PMD is primarily caused by 3 types of PLP1 mutations with different frequencies and clinical outcomes. PLP1 duplications are the most frequent cause of PMD, accounting for 50% to 75% of cases and typically resulting in the classic PMD phenotype.2,4,5 Point mutations represent another 20% of cases and lead to highly variable severities, ranging from mild to severe connatal disease, depending on whether the mutation alters the protein sequence (in exons) or disrupts splicing (in introns).3,5 Lastly, PLP1 deletions are the least common, found in less than 5% of cases; paradoxically, gene deletions are associated with milder phenotypes than other genetic changes in the PLP1 gene, such as duplications. 1
Predicting the clinical severity of PMD from a patient's specific PLP1 mutation is notoriously difficult because of significant clinical variability, even among members of the same family. Despite this challenge, several general genotype-phenotype correlations can be established. Point mutations that alter highly conserved amino acids typically lead to severe disease. Furthermore, disease severity is linked to gene dosage; having more than 2 copies of the PLP1 gene generally causes a more severe phenotype than a simple duplication (2 copies).6,7 In contrast, mutations that result in milder disease include those located within the PLP-specific exon 3B, as well as null mutations that cause a complete absence of PLP/DM20 protein, which often present with peripheral neuropathy. 3 These 4 cases from 2 families cover 2 of the most prevalent causes of PMD. Replication of the Xq22.1-q22.2 segment containing the OMIM gene PLP1 was noted in the 3 cases from the same family. This abnormality led to the development of classic PMD. The genetic test of the father was normal, but the genetic test of the mother could not be performed because of loss of contact, which is a limitation of this study. We assumed that the abnormal gene might have been passed on from the mother. However, the possibility of a gene mutation could not be ruled out. Regarding case 4, a c.737G>A missense mutation in the PLP1 gene was identified, resulting in G246E amino acid substitution. This variant was the cause of classic PMD in the case. Consistent with our observations, Goldman et al 1 classified this mutation as belonging to the classic PMD phenotype. His classification system categorizes forms 1, 2, and 3 under the umbrella of classic PMD. In contrast, Cailloux et al 3 classified the only other reported case of this mutation as form 1, a phenotype not identical to classic PMD. Despite the possibility of clinical heterogeneity, this discrepancy reflects the ambiguity within the current PMD classification system. Such a classificatory challenge is further highlighted by a different substitution at the same residue, G246A. This mutation was initially described as causing the mild form of PMD 8 but was later classified by Goldman et al 1 as connatal PMD.
The clinical severity of PMD caused by missense mutations is largely governed by the unfolded protein response (UPR) cellular stress pathway. The intensity of this response is dictated by how many PLP1 protein isoforms—that is, PLP and DM20—are retained in the endoplasmic reticulum (ER). Milder, classic PMD is often associated with mutations that trap only PLP, resulting in a weaker, protective UPR that promotes oligodendrocyte survival. In contrast, severe connatal PMD typically results from mutations that affect both PLP and DM20, inducing a strong, overwhelming UPR that leads to apoptosis and more severe disease. Although our model is powerful, our findings in case 4 challenge its predictive accuracy. The c.737G>A mutation is expected to impair the trafficking of both PLP and DM20, which should induce a strong UPR and a severe connatal phenotype. However, this patient presented with classic PMD. This discrepancy strongly suggests that the UPR is not the sole determinant of severity and that other factors, such as the efficiency of protein degradation pathways or the influence of genetic modifiers, also play a crucial role in modulating the final clinical outcome.9,10 Furthermore, the structural impact of the specific amino acid substitution must be considered. Residues of glycine, which is the smallest amino acid, are often critical for the tight packing of transmembrane helices. The substitution of glycine with glutamate (p.Gly246Glu) at position 246 introduces a large, charged side chain into a hydrophobic transmembrane domain. This results in significant steric hindrance and electrostatic repulsion, destabilizing the PLP1 protein structure. Although such severe structural disruption typically triggers a robust UPR and apoptosis leading to the connatal phenotype, the presentation of the classic form in our patient implies the occurrence of more complex intracellular processing. It is possible that a fraction of the mutant protein manages to escape the endoplasmic reticulum or that the specific conformational change is less cytotoxic than predicted. This highlights the limitation of relying solely on predictive models and emphasizes the necessity of functional studies to fully elucidate the pathogenicity of specific missense variants.
Having examined the pathogenic mechanism of the missense mutation in case 4, we now turn to the distinct genetic origins of the 2 mutation types presented in this study, duplications (cases 1-3) and point mutations (case 4). Chandley 11 noted a distinct parental bias for different types of de novo mutations. Most aneuploidies are of maternal origin, whereas point mutations and structural rearrangements (eg, duplications) are predominantly paternal. These differences stem from the distinct biological processes of oogenesis and spermatogenesis. However, in PMD, the parental origin of new mutations differs significantly by type. For PLP1 duplications, there is a strong male bias, as confirmed by a study by Mimault et al, 2 which found that 91% of mothers of affected sons were carriers. In sharp contrast, the same study showed no such bias for point mutations. The finding that 68% of mothers were carriers is consistent with an equal mutation rate between male and female germ cells, a scenario where one-third of new cases are expected to be de novo mutations. 2 Although the G246E mutation in the PLP1 gene was previously described in a case of maternal inheritance, 2 our article documents the first known instance of this specific mutation occurring de novo.
Although understanding these molecular mechanisms and genetic origins is critical for basic science, a more pressing challenge for clinicians is translating these complex findings into a timely and accurate diagnosis. The path to a definitive diagnosis of PMD is often a prolonged and difficult journey for families. A recent caregiver survey found that the average age at diagnosis was 4.5 years, with 25% of cases diagnosed before age 1 year. Despite the prevalence of genetic testing for PMD, a substantial diagnostic delay still occurs, with 70% of families reporting significant challenges throughout the process. 12 In our series, the ages at diagnosis for cases 1 and 4 were 15 and 13 months, respectively. Therefore, a systematic diagnostic algorithm is essential. The initial screening should target PLP1 gene duplications, as these are the most common cause of PMD. If this is unrevealing, sequencing for point mutations affecting the gene's exons and intronic splice sites should be conducted. 1 If testing for PLP1 variants is negative, a broad differential diagnosis, such as PMLD and other diseases with similar phenotypes, must be considered. Pelizaeus-Merzbacher-like disease 1 (PMLD1), an autosomal recessive disorder caused by mutations in the GJC2 gene (which encodes connexin 47), is the primary mimic, and it can sometimes be clinically indistinguishable from PMD. Mutations in GJC2 can also result in a milder phenotype, spastic paraplegia type 44 (SPG44). 13 However, it is notable that GJC2 mutations account for only about 8% of PMLD cases, 14 and many other hereditary leukodystrophies can also present with similar phenotypes. 15 Given this clinical overlap, caution is advisable when using the general term “PMLD,” and clinicians should look for specific clinical features to guide diagnosis. 1 Important distinguishing assessments include eye examinations for congenital cataracts, 16 dental examinations for hypodontia,17,18 and thyroid studies to rule out Allan-Herndon-Dudley syndrome. 18
The clinical presentation of PMD is characterized by nystagmus, ataxia, and developmental delay; these features significantly overlap with other hereditary leukodystrophies and neurocutaneous disorders, making accurate diagnosis difficult. Recent reviews emphasize the critical role of neuroimaging and clinical phenotyping in differentiating between these conditions.19,20 As summarized in Table 1, distinct patterns can be used to facilitate the specific diagnosis.
Differential Diagnosis of Pelizaeus-Merzbacher Disease, Other Leukodystrophies, and Neurocutaneous Disorders.
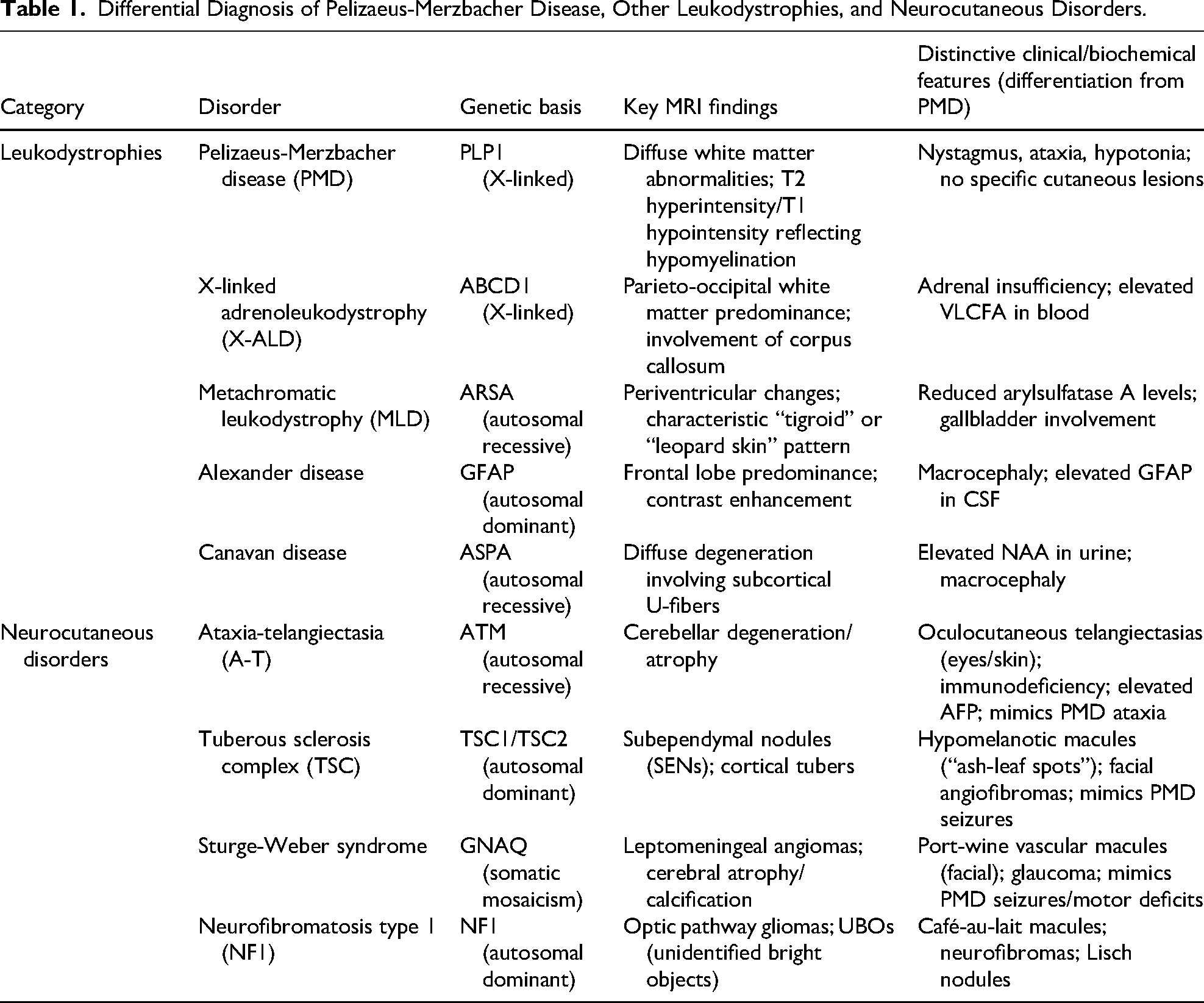
Although PMD is characterized by diffuse white matter abnormalities and a lack of apparent myelination on MRI, other leukodystrophies exhibit unique topographical distributions. For example, X-linked adrenoleukodystrophy typically manifests with parieto-occipital predominance, whereas Alexander disease exhibits frontal lobe dominance and Canavan disease involves subcortical U-fibers. 19 Furthermore, clinicians must be able to distinguish PMD from neurocutaneous syndromes (ie, phakomatoses) that present with similar neurologic sequelae. For example, ataxia-telangiectasia shares the hallmark symptom of progressive ataxia with PMD, but it can be clinically distinguished by the presence of oculocutaneous telangiectasias and immunodeficiency. 20 Similarly, although seizures may occur in PMD, their presence alongside cutaneous markers such as hypomelanotic macules or port-wine stains instead suggests tuberous sclerosis or Sturge-Weber syndrome, respectively, rather than PMD. 20 The absence of these cutaneous manifestations in patients with psychomotor regression should heighten suspicion for leukodystrophies such as PMD.
For affected families with a known history of PMD, prenatal diagnosis is possible. All types of PLP1 mutations, including point mutations, duplications, and deletions, can be detected using cells obtained from amniocentesis or chorionic villus sampling.1,21 Specific molecular techniques, such as quantitative fluorescence multiplex polymerase chain reaction or fluorescence in situ hybridization (FISH) probes, can be utilized for this analysis. 21 Furthermore, for families undergoing in vitro fertilization, preimplantation genetic diagnosis is also an option. 22
Once a definitive molecular diagnosis is established, whether prenatally or postnatally, management strategies and therapeutic options should be considered. However, there is currently no specific treatment for PMD. Medical care for PMD has long been limited to symptomatic treatments. 23 According to a survey conducted by Candice et al, 12 current management for PMD is supportive, with the most common interventions being physical therapy (100%), speech therapy (86%), and occupational therapy (81%). Regarding symptom progression over a year, no caregivers reported improvement; instead, the symptoms of 61% of patients were described as stable, whereas those of 34% of patients had worsened. Given the progressive nature of the disease, a proactive, multidisciplinary management approach is essential. Beyond standard physical and speech therapy, long-term care must address the secondary complications revealed in caregiver assessments, such as respiratory infections due to aspiration, nutritional deficits requiring gastrostomy, or orthopedic surgery for severe scoliosis or contractures. Although these supportive measures do not stop the underlying dysmyelination, they are vital for extending survival and improving the quality of life for both patients and their families. Meanwhile, first-in-class PMD therapies with novel mechanisms are currently under research. Strategies such as PLP1 suppression, modulating ER-stress-related sequelae, small molecules, and cell therapy bring hope to those suffering from PMD. 23
Specifically, ASO therapy has been proven effective in mice models 24 and is now being evaluated through a single-arm, open-label clinical trial whose success likely hinges on PLP1 knockdown efficiency. 25 Similarly, CRISPR-Cas9 gene suppression has shown dramatic, life-saving effects in the most severe PMD mouse models,26–28 although this approach remains in the preclinical stage because of challenges associated with delivery and the potential for off-target edits.24,29 Other strategies have targeted the downstream consequences of the mutation. Enhancing the trafficking of mutant PLP through cholesterol supplementation, for example, has shown promise in mouse models, 30 but has thus far failed to improve outcomes in a human clinical trial, 31 likely because of differences in the blood-brain barrier between the 2 species. Likewise, the UPR modulator curcumin showed only minimal benefit in animals, 32 and a subsequent human trial also failed to demonstrate clinical efficacy. 33 Another small molecule, the iron chelator deferiprone, was able to rescue cells in vitro, but it did not meaningfully alter the disease in mice. A related clinical trial is nonetheless being planned. Cell-based therapies aim to entirely replace defective oligodendrocytes. However, an early clinical trial using neural stem cells (HuCNS-SC) failed to show any benefit, 34 likely due to immune rejection of the transplanted cells. Hematopoietic stem cell transplantation (HSCT) has also been attempted, but its use is controversial because of a lack of clear scientific rationale for PMD, and case reports have shown its efficacy to be ambiguous. 35
Finally, the psychological and ethical dimensions of genetic counseling for PMD warrant specific attention. For families similar to the that of cases 1-3, where the mother could not be tested, the uncertainty regarding the carrier status of female relatives creates significant anxiety. Even when a de novo mutation is confirmed, as in case 4, parents often experience guilt or fear of recurrence in future pregnancies because of the theoretical risk of germline mosaicism. Therefore, genetic counseling should not be a one-time event, but instead an ongoing process. It must encompass not only risk assessment and reproductive options—such as prenatal diagnosis and preimplantation genetic testing—but also psychological support to help families cope with the emotional burden of raising a child with a chronic, debilitating disorder. Integrating professional psychological services into the standard care protocol for PMD families is as crucial as the physical rehabilitation of the patients. Future research and therapeutic development for PMD should be guided by the demands of the patient community. Caregivers have identified improved mobility and communication as their highest priorities. The most significant unmet needs include the ability to communicate, a therapy that could stop disease progression, and access to long-term care and financial support. Encouragingly, the PMD community appears highly motivated to participate in future research, as 69% of survey respondents indicated a willingness to enroll in a clinical trial or natural history study. 12
This study has several limitations that should be acknowledged. First, the small sample size, comprising only 4 patients from 2 families, restricts the generalizability of our findings to the wider population of individuals with PMD. A second limitation is the incomplete genetic analysis for the first family. The inability to perform genetic testing on the mother of cases 1-3 because of loss of contact prevents the definitive confirmation of the inheritance pattern. Although maternal inheritance is assumed, the possibility that the duplication was a de novo event cannot be definitively excluded. Third, the clinical data collected were not entirely uniform across all patients. Cases 2 and 3 did not undergo the same laboratory examinations as the other patients because of financial constraints. Furthermore, the follow-up assessments of patient progress were qualitative. This study would have been strengthened by the use of standardized developmental scales and motor function scores to objectively measure disease progression and the specific impact of rehabilitation therapy over time. Despite these limitations, this study provides a valuable clinical and genetic comparison of 2 distinct causes of classic PMD.
Conclusion
In summary, this article details 4 pediatric cases of classic Pelizaeus-Merzbacher disease, illustrating 2 of the most common genetic etiologies: a familial PLP1 gene duplication and a de novo missense mutation. Our findings reinforce the idea that clinicians should maintain a high index of suspicion for PMD in male children who present with the characteristic triad of nystagmus, hypotonia, and motor developmental delay. This study underscores the critical importance of a comprehensive genetic diagnostic approach. To ensure an early and accurate diagnosis, chromosomal copy number variation analysis should be performed in conjunction with gene sequencing, as PLP1 duplications are the leading cause of the disease. A definitive molecular diagnosis is essential not only for providing families with an accurate prognosis and crucial genetic counseling but also for guiding a holistic management strategy that integrates long-term multidisciplinary care with psychological support. Ultimately, these efforts pave the way for patient participation in future therapeutic trials.
Supplemental Material
Supplemental Material
Footnotes
Author Contributions
Haolin Chen and Chaonan Yu designed the study, collected and analyzed the data, and drafted the initial manuscript. Xiaoyi Fang and Yuanhong Ji supervised the study, acquired funding, and critically reviewed and revised the manuscript for important intellectual content. All authors read and approved the final manuscript. Haolin Chen and Chaonan Yu contributed equally to this work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the General Program of Basic Science Research of the Project of Natural Science Foundation of Shenzhen, 2025 (Grant No. JCYJ20250604143757075).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.