
Abstract
Extensive cocirculation of CRF01_AE and CRF07_BC in China has created favorable conditions for ongoing intersubtype recombination, contributing to increasing genetic complexity within the local HIV-1 epidemic. In this study, two novel unique recombinant forms composed of CRF01_AE and CRF07_BC were identified in Shenzhen, Guangdong Province. Near-full-length genome sequences were obtained for isolates LS11654 and LS16824. Phylogenetic analysis indicated that both sequences clustered within a CRF01_AE-/CRF07_BC-related lineage but were distinct from previously reported strains. Recombination analysis revealed markedly different mosaic structures: LS11654 contained two recombination breakpoints, whereas LS16824 exhibited a more complex genome with eight breakpoints and multiple inserted fragments. Subregion phylogenetic analysis further confirmed the parental origins of the recombinant segments. These findings reflect ongoing recombination driven by sustained cocirculation of CRF01_AE and CRF07_BC in Shenzhen and highlight the importance of continued molecular surveillance.
The global HIV epidemic remains a major public health challenge, largely driven by the virus’s extraordinary genetic variability. This diversity arises from the high error rate of reverse transcriptase, rapid viral turnover, and the highly recombinogenic nature of reverse transcription. Recombination between divergent strains can generate mosaic genomes known as circulating recombinant forms (CRFs), some of which subsequently establish regional epidemics and pose sustained challenges to prevention and treatment efforts.1,2 To date, recombination among HIV-1 group M subtypes has resulted in more than 170 designated CRFs and numerous unique recombinant forms (URFs), reflecting the ongoing diversification of the virus. 3
China’s HIV-1 epidemic is predominantly driven by CRFs, accounting for approximately 88% of reported infections nationwide. Among them, CRF07_BC (39.1%) and CRF01_AE (32.1%) represent the two most prevalent lineages, followed by CRF08_BC (9.2%) and emerging recombinants such as CRF55_01B (2.4%). 4 With multiple CRFs and subtypes cocirculating across regions over time, the proportion of URFs and other novel recombinants has shown a continuous upward trend in recent years. 5
The circulation of CRF01_AE in China is characterized by a complex multicluster transmission pattern. Multiple independent introductions during the late 1980s and 1990s led to the establishment of at least eight distinct transmission clusters, among which clusters 4 and 5 are predominantly associated with men who have sex with men (MSM) and have substantially shaped the MSM epidemic in China. 6 CRF07_BC was initially associated with people who inject drugs but has progressively expanded into MSM populations. Molecular epidemiological analyses have demonstrated extensive transmission clustering within MSM networks, highlighting its increasing contribution to the MSM epidemic. Currently, CRF07_BC is widely distributed across several major regions of China. 7 The cocirculation of CRF01_AE and CRF07_BC within MSM transmission networks provides favorable conditions for frequent recombination. In recent years, an increasing number of CRF01_AE-/CRF07_BC-related recombinants have been identified, including CRF79_0107 and CRF136_0107,8,9 as well as numerous newly reported URFs, reflecting ongoing genetic diversification.
Shenzhen, located at the Pearl River Estuary in Guangdong Province, is a core city within the Guangdong–Hong Kong–Macao Greater Bay Area and is characterized by a highly mobile population. Its substantial migrant population creates a dynamic demographic structure that may facilitate viral transmission and diversification.
Recent molecular surveillance has revealed considerable genetic diversity in the local HIV-1 epidemic. CRF07_BC remains the predominant genotype, followed by CRF01_AE, CRF55_01B, subtype B, CRF08_BC, and multiple URFs. Notably, the proportions of CRF07_BC, CRF55_01B, and URFs have shown a gradual upward trend in recent years. CRF01_AE has established relatively independent and active transmission networks in Shenzhen, with its spread closely associated with large-scale population mobility.10,11
MSM constitute the primary affected population, accounting for over half of newly reported infections, and exhibit highly active molecular transmission clusters. Meanwhile, heterosexual transmission remains substantial, and increasing infections among younger individuals and older adults suggest potential expansion of the epidemic into broader population groups. 12
Against this background of high genetic diversity, active transmission networks, and increasing recombinant complexity, an in-depth investigation of HIV-1 genomic variation, particularly the underexplored URFs, is essential for understanding local viral evolution and informing precision prevention strategies in Shenzhen and southern China.
In this study, two previously unreported HIV-1 URFs, each comprising genomic regions derived from CRF01_AE and CRF07_BC, were identified in plasma specimens collected from two epidemiologically unrelated individuals (LS11654 and LS16824) in Shenzhen, southern China.
Case LS11654 was a 23-year-old unmarried male infected through heterosexual contact, with a plasma sample collected on May 15, 2014. Case LS16824 was a 36-year-old unmarried man who acquired HIV through male-to-male sexual contact, with a sample collected on October 15, 2015. This study was conducted as part of routine HIV molecular surveillance in Shenzhen, China. Written informed consent was obtained from all participants prior to sample collection.
Viral RNA was extracted from 280 μL of plasma using the QIAamp Viral RNA Mini Kit (Qiagen, Germany). Reverse transcription was performed using the SuperScript™ IV First-Strand Synthesis System (Thermo Fisher Scientific, USA) to generate two overlapping half-genome cDNA fragments.
Nested PCR amplification was carried out using PrimeSTAR® Max DNA Polymerase Ver.2 and PrimeSTAR GXL Premix Fast (Takara Bio, Japan). Amplified products were examined by agarose gel electrophoresis, followed by purification with the TIANquick N96 kit (Tiangen Biotech, China) and DNA quantification using the Quant-iT™ PicoGreen™ dsDNA assay (Thermo Fisher Scientific).
Equimolar amounts of purified amplicons were pooled and sequenced on the PacBio Revio platform (Tianyi Huiyuan Life Science and Technology Co., Beijing, China). Raw sequencing reads were quality filtered and assembled into contiguous consensus sequences using ContigExpress (version 11.5.1).
The obtained near-full-length genome (NFLG) sequences were analyzed using the Basic Local Alignment Search Tool against the HIV sequence database. No highly similar sequences (>95% nucleotide identity) were identified.
The two NFLG sequences were aligned with a curated panel of HIV-1 reference sequences using MAFFT v7.487. The reference dataset included one HIV-1 group O sequence, two representative sequences from each major group M subtype, and multiple representative CRFs prevalent in China, including CRF01_AE, CRF07_BC, CRF08_BC, and CRF55_01B. Representative sequences of known CRF01_AE/CRF07_BC recombinants were also included. All reference sequences were retrieved from the HIV Database. 13
The multiple sequence alignment was manually edited in BioEdit (version 7.2.5.0) to optimize alignment quality.
Phylogenetic relationships were reconstructed using the maximum-likelihood approach implemented in FastTree v2.1.8 with the General Time-Reversible model (GTR) + discrete rate categories approximation for among-site rate heterogeneity (CAT) substitution model. Topological refinement was performed with four rounds of subtree-prune-regraft (with a value of 4), and the nodal support was assessed using the Shimodaira–Hasegawa (SH)-like local support method.
Recombination analysis was initially performed using the Recombinant Identification Program (RIP), available at HIV Database (https://www.hiv.lanl.gov/content/sequence/RIP/RIP.html). The identified recombinant breakpoints were further examined and confirmed using bootscanning analysis implemented in SimPlot version 3.5.1. The recombinant genome structures were subsequently visualized using the Recombinant HIV-1 Drawing Tool available at the Los Alamos HIV database (https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html).
To confirm the parental origin of each recombinant fragment, phylogenetic analyses were performed for individual subregions. Corresponding genomic segments were extracted and aligned separately with reference sequences. Neighbor-joining phylogenies were generated using MEGA X with the Kimura two-parameter substitution model, and statistical support for internal branches was determined through 1,000 bootstrap iterations.
NFLG sequences of 8,816 bp for LS16824 and 8,840 bp for LS11654 were successfully obtained, spanning nucleotide positions 790–9,411 relative to the HXB2 reference strain.
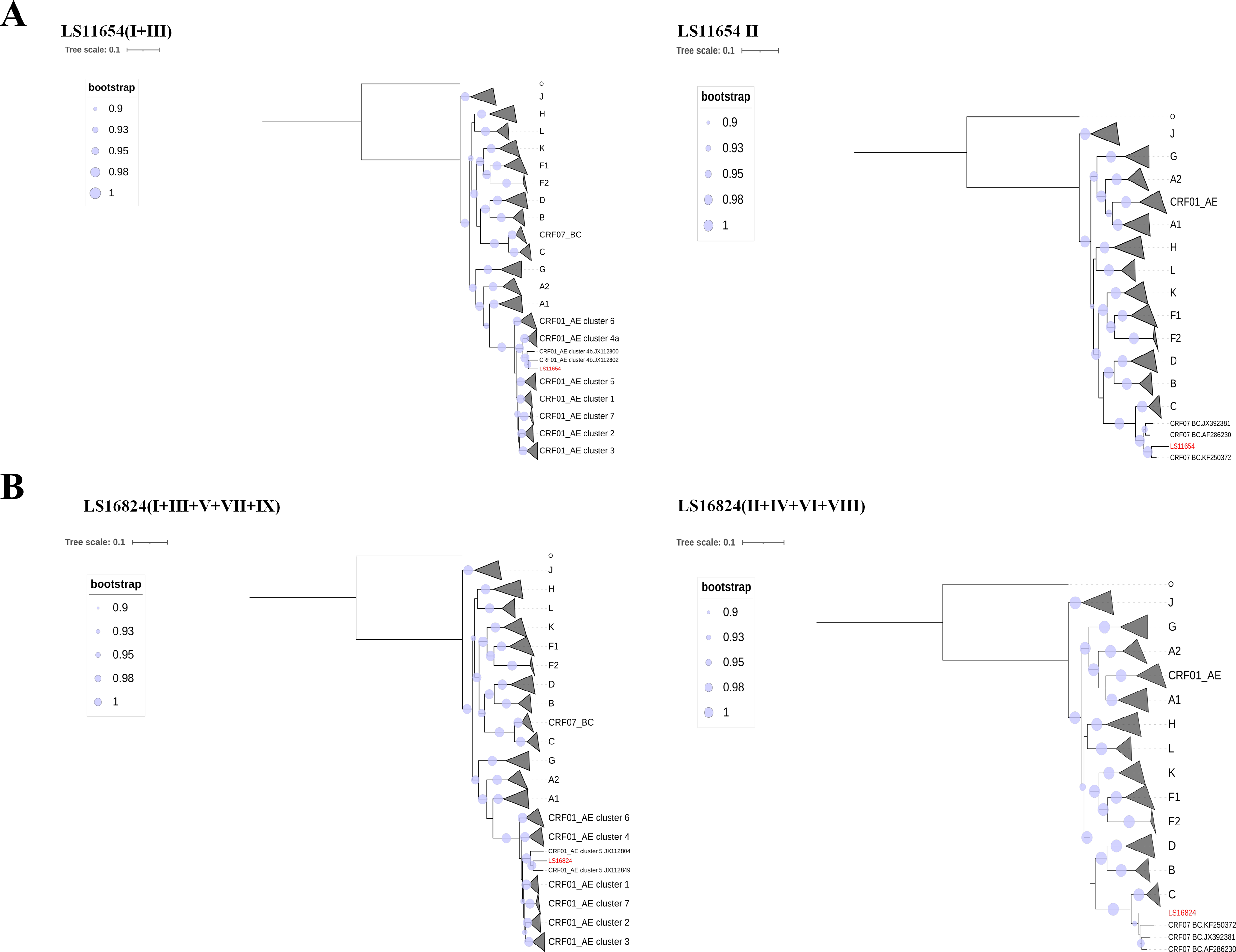
As shown in Figure 1, maximum-likelihood phylogenetic analysis based on NFLG sequences demonstrated that LS11654 and LS16824 clustered within the CRF01_AE/CRF07_BC recombinant lineage with strong support (SH-like support of >0.98). However, neither sequence formed a tight monophyletic cluster with any previously reported strains; instead, both occupied distinct peripheral positions in the phylogenetic tree, suggesting that they represent novel recombinant forms.
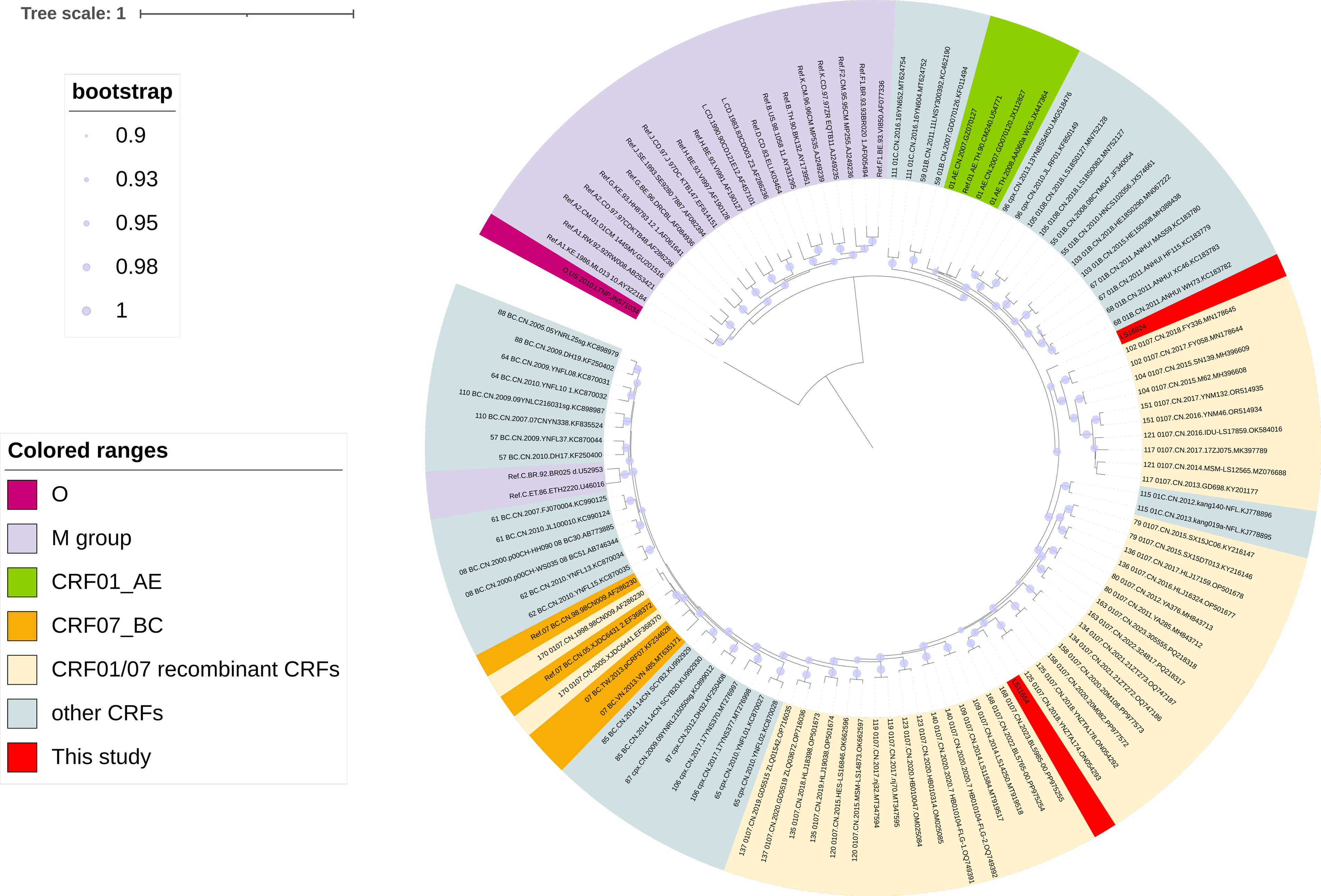
Maximum-likelihood phylogenetic tree based on near-full-length genome (NFLG) sequences. The tree was reconstructed using FastTree v2.1.8 under the GTR + CAT approximation model with four rounds of subtree-prune-regraft optimization (SPR = 4). Branch support was assessed using Shimodaira–Hasegawa (SH)-like local support values. SH-like support values ≥0.90 are indicated by filled circles at the corresponding nodes. Colored outer bands indicate major HIV-1 groups, subtypes, and circulating recombinant forms (CRFs). Sequences generated in this study (LS11654 and LS16824) are highlighted in red. Branch lengths represent nucleotide substitutions per site. CRF, circulating recombinant form; NFLG, near-full-length genome.
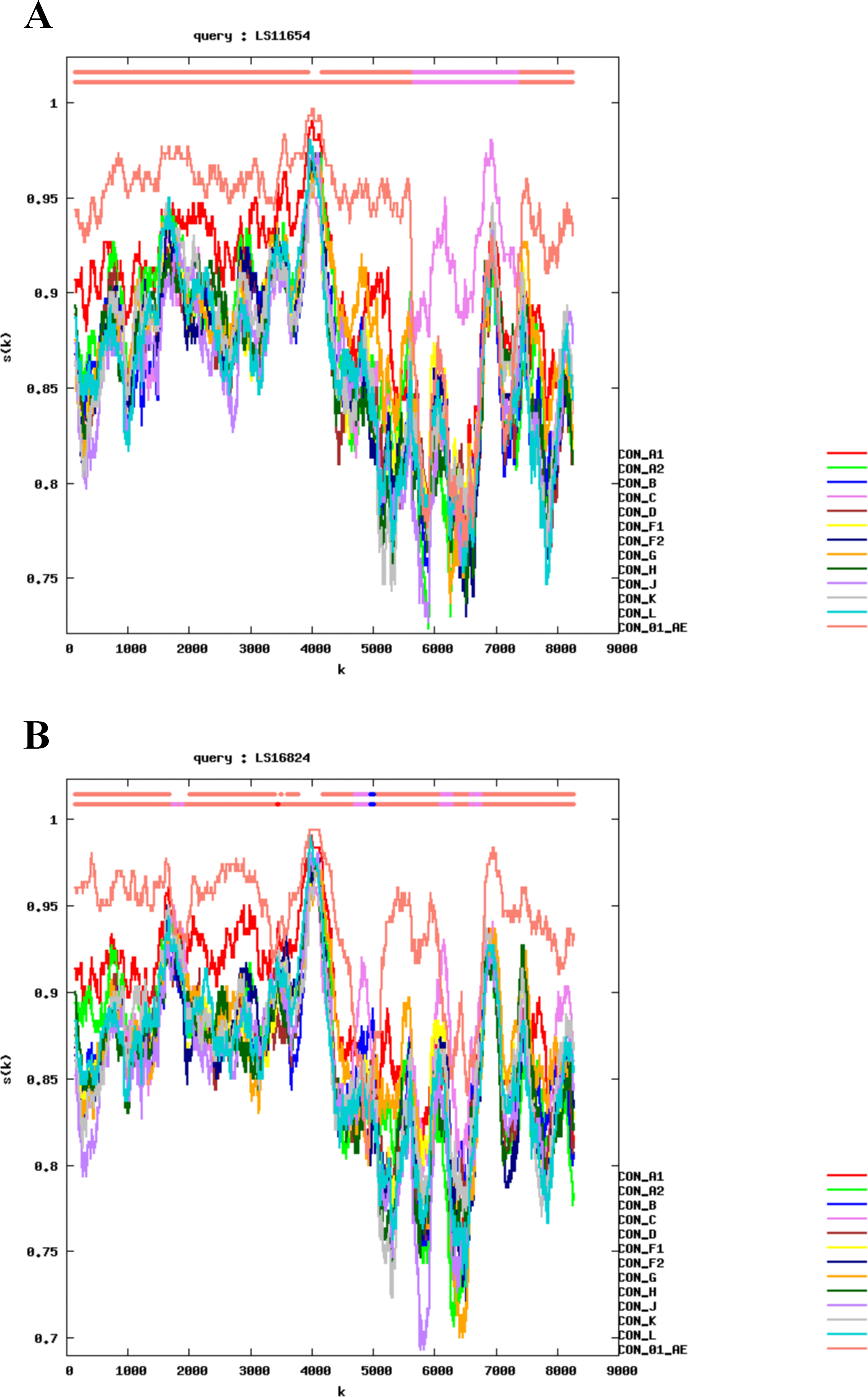
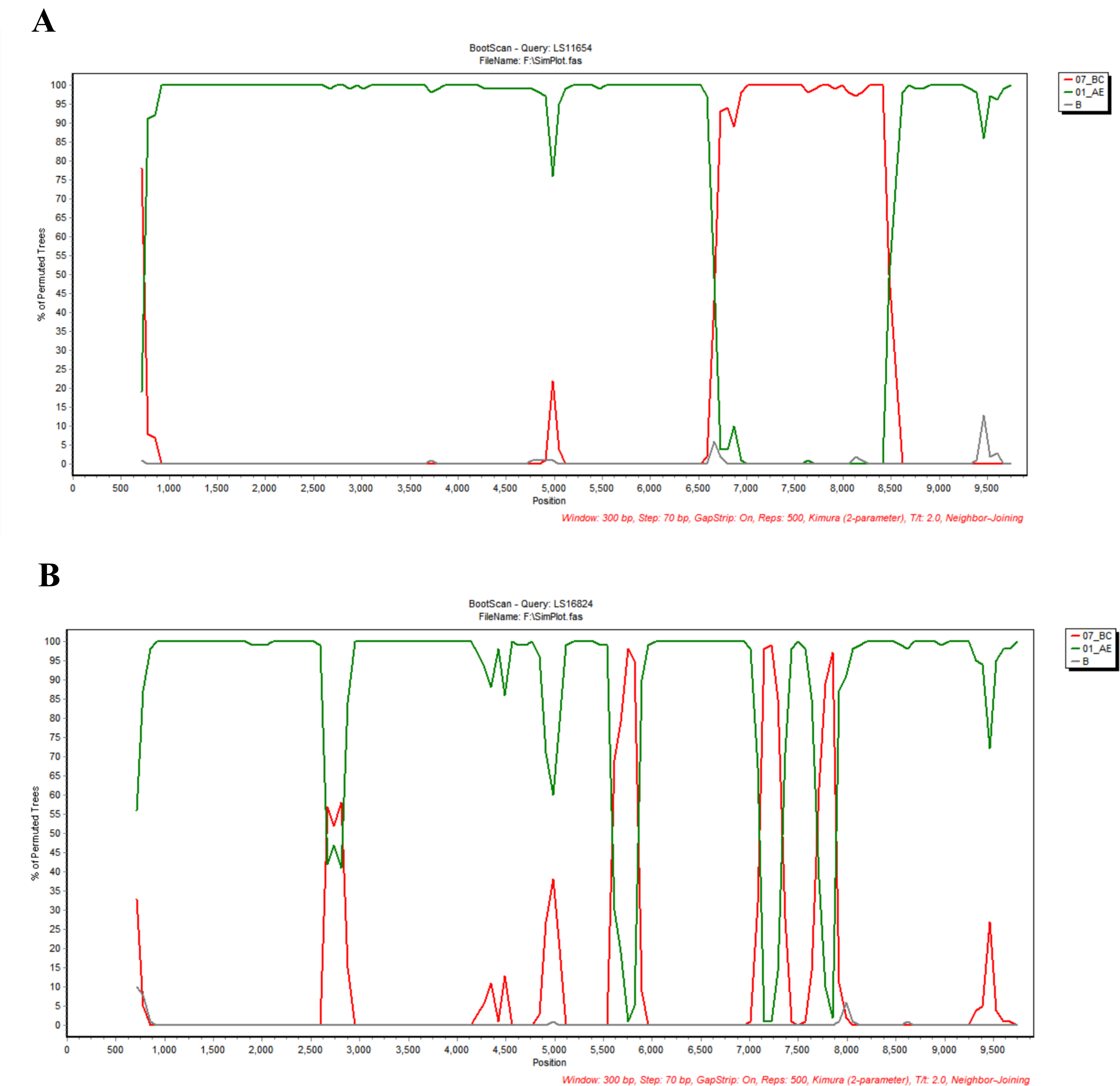
Recombination analysis using RIP and bootscanning analysis demonstrated that both NFLG sequences exhibited mosaic genome structures composed of CRF01_AE and CRF07_BC segments (Figs. 2 and 3). The detailed recombinant structures and breakpoint distributions were further illustrated using the HIV-1 Drawing Tool (Fig. 4).
Recombinant Identification Program (RIP) analysis of the NFLG sequences.
Bootscan analysis of NFLG sequences.
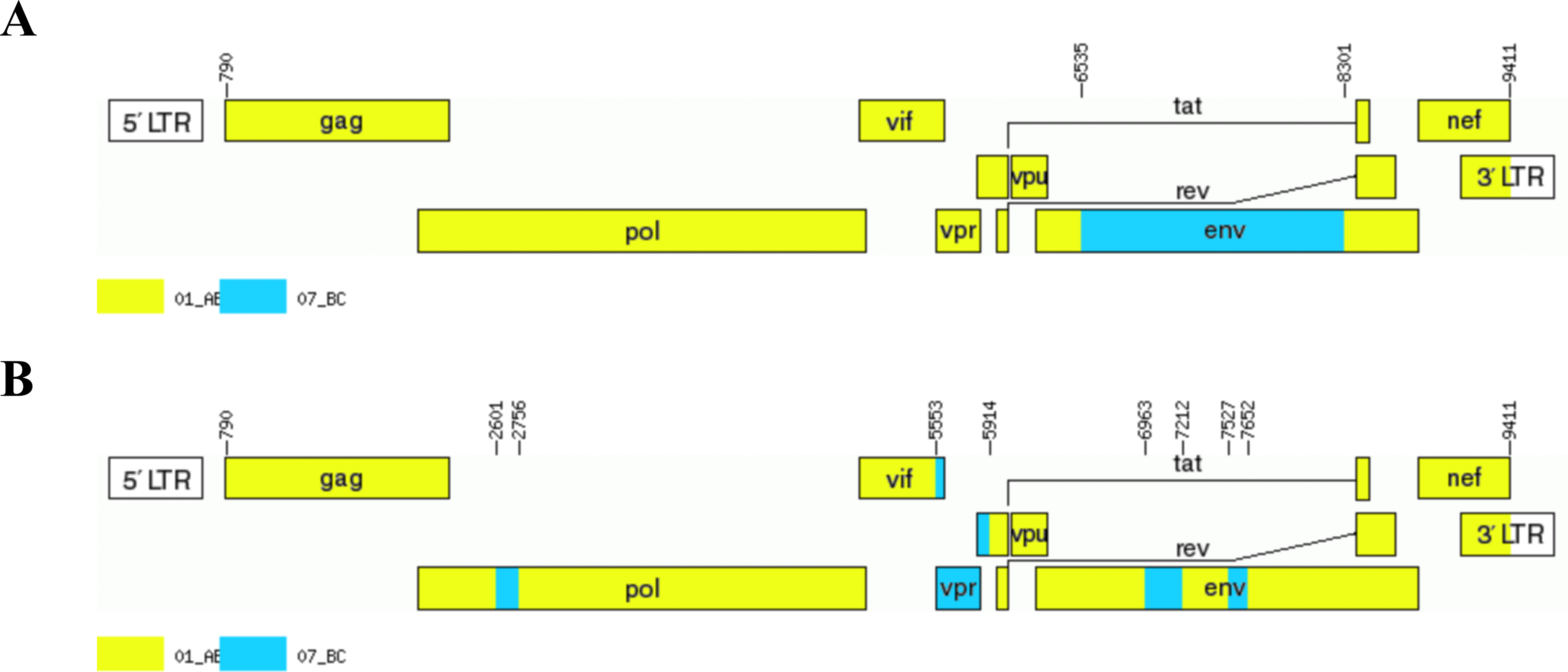
Genetic mosaic structures of NFLG sequences.
The recombinant genome of LS11654 consisted of three segments separated by two recombination breakpoints. Using CRF01_AE as the backbone, a CRF07_BC-derived fragment was inserted within the env region. Specifically, segment I corresponded to CRF01_AE (790–6,535 nt), segment II to CRF07_BC (6,536–8,301 nt), and segment III to CRF01_AE (8,302–9,411 nt).
In contrast, the recombinant genome of LS16824 displayed a more complex mosaic structure comprising nine segments separated by eight recombination breakpoints. With CRF01_AE as the backbone, multiple CRF07_BC-derived fragments were inserted within the pol, vif, vpr, and env regions. The genomic composition was as follows: segment I, CRF01_AE (790–2,601 nt); segment II, CRF07_BC (2,602–2,756 nt); segment III, CRF01_AE (2,757–5,553 nt); segment IV, CRF07_BC (5,554–5,914 nt); segment V, CRF01_AE (5,915–6,963 nt); segment VI, CRF07_BC (6,964–7,212 nt); segment VII, CRF01_AE (7,213–7,527 nt); segment VIII, CRF07_BC (7,528–7,652 nt); and segment IX, CRF01_AE (7,653–9,411 nt).
Subregion phylogenetic analyses further confirmed the parental origins of the recombinant fragments (Fig. 5). The CRF01_AE-derived fragment of LS11654 clustered with CRF01_AE reference sequences and showed a close phylogenetic relationship with cluster 4, with strong bootstrap support (100%). The CRF07_BC-derived fragment clustered with CRF07_BC reference sequences and with strong bootstrap support (100%).
Subregion phylogenetic analyses of recombinant fragments from LS11654
Similarly, the CRF01_AE-derived fragment of LS16824 clustered with CRF01_AE reference sequences and was closely related to cluster 5, with strong bootstrap support (100%). The CRF07_BC-derived fragment clustered with CRF07_BC reference sequences, supported by a bootstrap value of 91%.
Shenzhen, a major gateway city in southern China, is characterized by a highly mobile migrant population and substantial demographic turnover. Such features facilitate the introduction and cocirculation of diverse HIV-1 genotypes, creating favorable conditions for intersubtype recombination. In this context, CRF01_AE and CRF07_BC remain the predominant circulating strains in the region.
In this study, two CRF01_AE/CRF07_BC URFs were identified in Shenzhen, further confirming the cocirculation of these two major CRFs locally. 14 The detection of these URFs suggests that co-infection or superinfection with distinct CRFs may occur within local transmission networks, thereby providing opportunities for ongoing recombination and contributing to increasing recombinant diversity.
Subregion phylogenetic analysis revealed that the CRF01_AE fragment of LS11654 clustered within cluster 4, whereas that of LS16824 clustered within cluster 5, both of which are CRF01_AE transmission clusters predominantly associated with MSM in China. Although LS16824 acquired HIV through male-to-male sexual contact, LS11654 acquired infection through heterosexual contact. These findings indicate that CRF01_AE/CRF07_BC recombination is not confined exclusively to MSM transmission networks but may also occur across different transmission groups, reflecting the interconnected nature of local epidemic networks. 15
Notably, the two URFs exhibited markedly different mosaic structures. LS11654 displayed a relatively simple recombinant pattern comprising three segments, whereas LS16824 harbored a more complex mosaic genome with nine segments and multiple recombination breakpoints. The presence of distinct recombination patterns in two independent individuals suggests that CRF01_AE/CRF07_BC recombination events are not isolated occurrences but may reflect active and continuous recombination processes within local transmission networks. The increasing structural complexity observed in some recombinants may also indicate successive or second-generation recombination events in settings where multiple CRFs cocirculate.
Taken together, the identification of these two URFs underscores the increasing genetic complexity of HIV-1 strains circulating in Shenzhen. Continuous molecular surveillance across diverse populations is therefore essential to better understand local transmission dynamics and to monitor the potential emergence of new recombinant forms.
Sequences Data
The gene sequences of LS16824 and LS11654 have been deposited in GenBank under accession numbers PX938385 and PX938386, respectively.
Authors’ Contributions
C.L., J.H., H.W., and L.L. designed the study. C.L. performed the experiments. C.L., B.Z., and D.L. participated in data analysis. All authors contributed to the writing and revision of the article and approved the final version.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the