
Abstract
Neurogenic bowel (NB) affects roughly 60% of people with a spinal cord injury (SCI), and these patients present with slow colonic transit, constipation, and chronic abdominal pain. The mechanisms by which NB bowel develops are unclear, thereby limiting interventions to being primarily symptom-focused and ineffective. Therefore, the main goal of this study was to identify the mechanisms that initiate and maintain NB after SCI as a critical step to develop evidence-based, novel therapeutic options to prevent NB. In previous studies, the neurogenic inflammatory mediator calcitonin gene-related peptide (CGRP) was identified as a high-priority candidate gene. Therefore, in a midthoracic rodent spinal contusion model that presents with clinically translatable NB-like phenotypes, we conducted intrarectal antagonism of CGRP activity using CGRP8–37 (compared to vehicle administration) in mice with SCI. This was followed by histological, molecular, and functional (Ca2+ imaging) approaches to assess the prevention of previously reported phenotypes of NB. CGRP8–37 significantly prevented colonic dysmotility and structural defects of the colon (i.e., expanded lymphoid nodules). There was also a prevention of microbial invasion into the colon wall and neuronal hyperresponsiveness to autologous fecal supernatants. These data support the role of CGRP/CGRP as a candidate mechanism for NB after SCI and highlight the potential for novel therapeutic treatments for the prevention of NB.
Keywords
Introduction
Neurogenic bowel (NB), characterized by slow colonic transit, fecal incontinence, and persistent abdominal pain,1–5 affects the majority of people with chronic diseases of the central nervous system (CNS), including multiple sclerosis, Parkinson’s disease, and spinal cord injury (SCI). 6 Up to 95% of patients with SCI (PWSCI) report delayed colonic transit7–10 and constipation with ∼75% experiencing fecal incontinence due to NB.11,12 In addition, despite loss of normal sensory function below the level of spinal injury, abdominal pain is experienced by 80–90% of all patients. 13 NB rarely resolves and escalates over time, making it a long-term physical and psychological challenge for PWSCI, their caregivers, and physicians. Once patients transition to the rehabilitative phase following injury, NB treatment is primarily focused on achieving fecal continence and avoiding constipation through a multipronged approach to bowel management, but the lack of uniform guidelines based on strong supporting evidence means that trial-and-error plays a significant role in the development of individualized bowel management programs.11,14,15 The knowledge gap around the mechanisms underlying bowel pain and dysfunction after SCI remains a significant barrier to the development of effective treatments.
While the loss of sensory and motor function after SCI is clearly linked to neuronal damage in the CNS, recent evidence suggests that the peripheral nervous system plays a critical role in the development of pain after spinal injury. Primary afferents innervating the skin and muscle of the hindlimbs contribute to enhanced transmission of pain signals after SCI,16–19 but their potential role in the development of NB is not known, even though pain is a primary NB symptom. Our recent characterization of an NB-like phenotype in mice with SCI closely aligns with the clinical presentation of NB, 20 providing an opportunity for investigation into tissue-specific mechanisms playing a role in the development of NB. Briefly, we identified an increased abundance of Calca and Calcb (i.e., the two transcript isoforms encoding calcitonin gene-related peptide [CGRP], a neurogenic inflammatory mediator) at 1 and 7 days after SCI, respectively. Expression is almost exclusively restricted to extrinsic primary afferents (Calca; spinal and vagal) and intrinsic afferents of the enteric nervous system (Calcb), suggesting that extrinsic CGRP synthesis/release precedes that of intrinsic, in line with evidence of elevated CGRP in the skin after SCI. 19 This highlights a potential feedforward loop in the bowel whereby extrinsic afferent-mediated neurogenic inflammation is amplified and perpetuated by intrinsic neurons of the enteric nervous system (ENS).
Under normal conditions, the ENS seems to play a minimal role in the transmission of bowel pain, but it is unclear what role interactions between intrinsic and extrinsic visceral innervation may play under pathological conditions (e.g., after injury or inflammation). 21 Along these lines, calcium (Ca2+) imaging of ENS neurons and colon-specific vagal afferents confirmed a trajectory of neuronal hyperresponsiveness to multiple stimuli, including fecal supernatants after SCI, providing a unique cellular path for pain signals to reach the brain while bypassing spinal processing, which may be disrupted as a result of injury.
CGRP overexpression corresponds to the onset of SCI-induced dysmotility, expansion of nearby lymphatic nodules within the colon, and facilitation of bacterial translocation from the gut lumen into the colon wall. 20 We hypothesized that a neurogenic inflammatory process initiated by CGRP synthesis and release from extrinsic primary afferents was responsible for the development of NB. The present studies test whether intrarectal administration of a CGRP antagonist (CGRP8–37) could prevent the development of NB-like phenotypes after SCI.
Methods
Mice
Adult (8–10 weeks old) female C57BL/6J mice from Jackson Labs (Bar Harbor, ME, USA) were used for this study. Males were excluded based on data indicating reduced bladder voiding efficacy following SCI compared to females, which could increase mortality. However, clinical reports of similar neurogenic 22 bowel dysfunction scores after spinal injury 23 and comparable effectiveness of FDA-approved CGRP antagonists in females and males, 24 particularly when used as a preventative rather than a treatment for acute migraine, point to the likelihood that intervention-related findings could generalize to both sexes. All mice were housed at the University of Kansas Medical Center on a 12:12 light:dark cycle with food and water available ad libitum and a fiber nestlet available. Animal use protocols conformed to NIH guidelines and were approved by the Institutional Animal Care and Use Committees at the University of Kansas Medical Center and conformed to the Committee for Research and Ethical Issues of IASP.
Spinal contusion injury
Mice were randomly sorted into control or treatment groups and then anesthetized with isoflurane (Dechra Veterinary Products, Overland Park, KS, USA) at 5% induction and 2% maintenance. A moderate severity contusion injury at T9 (65 kDynes of force with one second dwell time) was performed with IH-0400 Infinite Horizons Impactor (Precision Systems and Instrumentation, Fairfax Station, VA, USA) as described previously 25 followed by intrarectal instillation with a vehicle control or CGRP8–37 (described in the next section). Injured mice were returned to their home cages to recover and received daily 5 mg kg−1 of gentamicin sulfate via intraperitoneal (i.p.) injections for 5 days to prevent postsurgical infection. 26 Rodents were assessed at day 1 (D1), day 7 (D7), and day 28 (D28) postinjury.
Drug dosage and delivery
CGRP8–37 (Cayman Chemical Co., Ann Arbor, MI, USA) dose was determined based on prior reports of the impact of CGRP8–37 on hyperalgesic priming, which supported the role of CGRP in pain processing. 27 Paige et al. administered CGRP8–37 intrathecally (1 μg in 5 μL of 1x phosphate-buffered saline [PBS]), so to adapt this dosage for intrarectal (i.r.) administration, we multiplied both the dosage and volume by 10. Mice in the vehicle control group received an intrarectal instillation of 50 μL of 1x PBS, while the treatment group was instilled with 10 μg of CGRP8–37, resuspended in 50 μL of 1x PBS.
Basso Mouse Scale
The Basso Mouse Scale (BMS) for locomotion defects was conducted as described. 28 Mice with an SCI were individually placed on a flat surface and allowed to freely move for 4 min. Motor function was assessed using the scale for each hind leg, and the left and right hind leg scores were averaged for the overall mouse score.
Colonic motility
Colonic transit was measured using a bead assay as previously described. 29 Mice were anesthetized using isoflurane (5% induction, 2% maintenance), and a 2 mm diameter glass bead (Sigma-Aldrich, Saint Louis, MO, USA) was gently pushed intrarectally 3 cm into the distal colon using a fistula probe (Roboz Surgical Instrument Co., Inc., Gaithersburg, MD, USA). A stopwatch was immediately started, and colonic transit time (CTT) was calculated based on latency to bead expulsion.
Fecal water weight assessment
Four fecal pellets from each mouse were weighed in grams (g) to four decimal places. The pellets were then microwaved for 6 min at power level four in a microwave (60 Hz, 1500 W, 2450 MHz). Following dehydration, the pellets were weighed again. The wet weight and dry weight were normalized by dividing each weight by the number of pellets. The difference between the normalized wet and dry weights represents the water content (g) and was used in all statistical analyses.
Histology and fluorescent in situ hybridization
Roughly 4.5 cm of the distal colon was postfixed in 10% neutralized formalin (Sigma-Aldrich, Saint Louis, MO, USA). After 24 h, the fixed colons were paraffin-embedded and cut into 8 μm thick cross-sections using a microtome. Slides were stained for goblet cells and mucins using an Alcian blue dye and Nuclear Red Fast counterstain kit (Abcam, Cambridge, UK). Quantification of the percent area of Alcian blue staining was calculated using the Alcian blue color deconvolution plugin in FIJI. 30 All images were taken using a Nikon 80i microscope (Nikon Instruments, Inc., Melville, NY, USA).
Fluorescent in situ hybridization (FISH) for localization of bacteria in the intestinal lumen was conducted as previously described. 31 After dewaxing and rehydrating tissue using a ClearRite-ethanol gradient, slides were stained using 5 μg mL−1 of EUB338 probe conjugated with a TEX615 fluorophore (Integrated DNA Technologies, Coralville, IA, USA) for fluorescent microscopy visualization. Slides were counterstained with Vector Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Inc., Newark, CA, USA). All images were taken using a Nikon 80i microscope. For each FISH prepared section, the sterile distance between the luminal-facing, mucosal epithelial layer of the colon wall (DAPI stain) and the outermost border of gut lumen contents (EUB338 probe) was visualized. In order to quantify the sterile distance and account for variations in section size and shape, the first distance marker for each section was placed in the lower center portion of the section. For each subsequent location (the remaining 9/10), the experimenter placed markers at approximately equal intervals around the internal circumference of the section. The length of each line was quantified using FIJI. The mean value of all 10 measures was then used for all statistical analyses.
Single-cell gel electrophoresis comet assay
Double-stranded DNA breaks from colon samples were quantified using the OxiSelectTM Comet Assay Kit (3 Well Slides), also known as single-cell gel electrophoresis (Cell BioLabs, Inc., San Diego, CA, USA), according to the manufacturer’s instructions. DNA “comets” were visualized on a Nikon 80i microscope, and images captured at 10X magnification. 20 DNA comets were analyzed per mouse. The images were blinded, and two independent experimenters rated all comet tails on a scale from 1 to 5, with one representing little to no comet tail and five representing a full tail, as previously described. 32 Experimenter scores were averaged for all comets, and this average was used for all subsequent analyses.
Retrograde labeling of colon-specific, extrinsic primary afferents
Retrograde labeling was carried out 7 days before sacrifice. Mice were anesthetized with gaseous isoflurane (5% induction, 2% maintenance), and a laparotomy was conducted to open the abdominal cavity. Each mouse received a series of injections of Alexa Fluor 488-conjugated cholera toxin B (CTB) (Invitrogen, Waltham, MA, USA) at a concentration of 1 mg mL−1. These injections were inserted into the subserosa of the distal colon using a 1-inch, 33-gauge Hamilton needle with an angle of 12 and point style of 4 (Hamilton Company, Reno, NV, USA). The cumulation of the injections’ total volume was 10 μL per mouse, and then the abdominal wall and skin were sutured for healing.
Ca2+ imaging
Ca2+ imaging was conducted as previously described.33–35
Bilateral nodose ganglion (ND) sensory neurons of the vagus nerve, as well as the myenteric plexus isolated from the intestinal wall, were removed and dissociated as previously described.36–38
All neuronal subpopulations were plated onto 12-mm poly-
Following FURA incubation, the cover-slips were mounted on an inverted Nikon TiE microscope stage (Nikon Instruments, Inc., Melville, NY, USA) with a pco.edge 4.2 LT USB sCMOS camera (Excelitas Technologies Co., Waltham, MA, USA). 1x HBSS constantly flowed over the cover-slip at 5 mL min−1, controlled by a gravity flow system (Warner Instruments, Holliston, MA, USA). Cells were treated with agonists through a gravity-feed pinch valve control perfusion system (Warner Instruments, Holliston, MA, USA). The cells were excited by a 175 W Xenon illumination system with a high-speed filter wheel (Sutter Instrument Co., Novato, CA, USA), and responses were measured as the ratio of 340/380 nM excitation and 510 nM emission (ΔF340/380). Cells were briefly (5 sec) exposed to 30 mM K+ solution (high K+). Response to high K+ was used as criteria for identification of cells as “living” with a peak Ca2+ influx (ΔF) cutoff of at least 0.1. Five minutes after high K+, cells were then briefly stimulated with fecal supernatants to determine cellular responses.
Fecal supernatants were created by removing three pellets of fresh feces from a rodent at the time of sacrifice and stored at −20°C until use (no more than 3 months). The morning of Ca2+ imaging, the three fecal pellets were homogenized in 1 mL of colorless 1x HBSS, centrifuged for 30 sec to pellet the fecal material, and the remaining supernatant was diluted 1:1 with colorless 1x HBSS for a final working solution. During imaging, 75 μL of the fecal supernatant was carefully pipetted onto the cover-slip. In postimaging analysis, cells were sorted by the presence of CTB for calculation of peak Ca2+ influx (ΔF), and the proportion of cells responding to each agonist was calculated.
Statistical analyses
All statistics were calculated using Statistical Package for the Social Sciences (SPSS v. 27, IBM, Armonk, NY, USA). All data are presented as mean ± standard error of the mean. All behavioral data were analyzed using a mixed design, two-way repeated measures univariate analysis of variance (ANOVA) to assess the effect of treatment and time on the dependent variables tested. For experiments with significant main effects and/or interactions, follow-up independent samples t-tests without Bonferroni correction were conducted as post hoc analyses for planned comparisons of vehicle versus CGRP8–37 within each time point. We statistically analyzed the datasets reporting the Alcian blue stain, comet assay, EUB338 stain, and ΔF in Ca2+ imaging using a two-way ANOVA followed by independent samples t-tests without Bonferroni correction within each timepoint. Finally, for the proportion of cell response in Ca2+ imaging, we conducted chi-squared tests to assess for differences between conditions and treatment, followed by individual chi-squared post hoc tests without Bonferroni correction within each time point. A p < 0.05 was considered significant in all analyses, as indicated by the * symbol.
Results
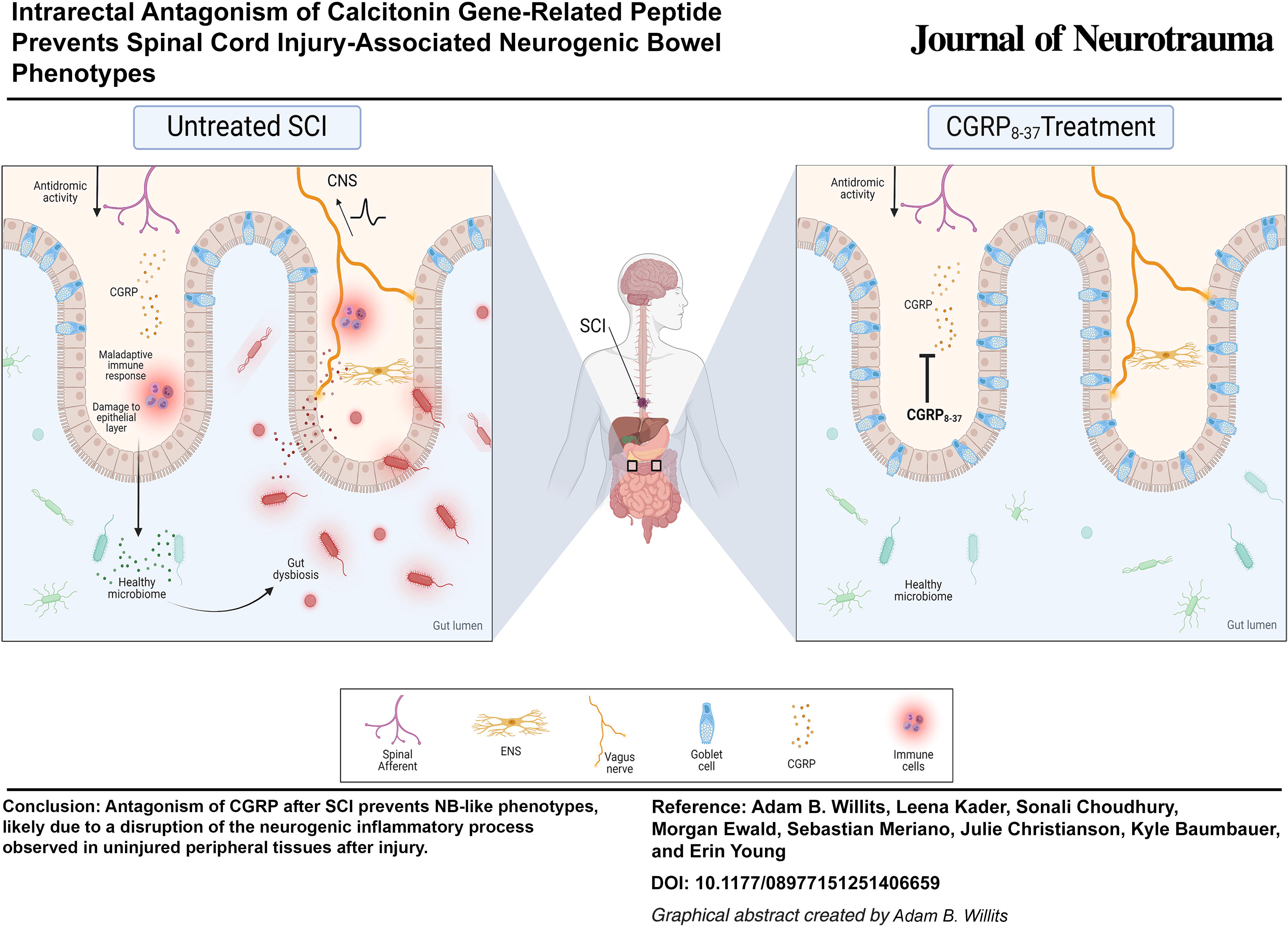
CGRP8–37 prevents colonic dysmotility but does not affect BMS, body weight, or fecal water composition
Both vehicle and CGRP8–37-treated groups exhibited significant changes in body weight over time, but no significant effect of treatment was observed (Fig. 1A). A 2 (treatment) × 9 (time) mixed design repeated measures ANOVA confirmed a significant main effect of time (F[8,136] = 32.087, p < 0.001) on body weight. No other significant main effects or interactions (all F < 1.083, all p > 0.05) were observed. Similarly, both groups showed deficits in locomotor activity after SCI with an improvement in BMS score over time. CGRP8–37 treatment had no effect on BMS (Fig. 1B). A 2 (treatment) × 3 (time) mixed design repeated measures ANOVA confirmed a significant main effect of time (F[2,34] = 48.272, p < 0.001) on BMS but no other significant main effects or interactions (all F < 1.083, all p > 0.05).
CGRP8–37 significantly prevents colonic transit defects after SCI.
CTT was significantly increased in vehicle-treated mice, and CGRP8–37 treatment prevented this phenotype (Fig. 1C). A 2 (treatment) × 4 (time) mixed design repeated measures ANOVA confirmed a significant main effect of treatment (F[1,17] = 8.988, p = 0.008) and time (F[3,51] = 7.313, p < 0.001) as well as a significant treatment × time interaction (F[3,51] = 5.243, p = 0.003). Follow-up planned comparisons with independent samples t-tests were conducted to compare vehicle and CGRP8–37-treated mice at each time point. We observed no significant difference between vehicle and CGRP8–37 for preinjury baseline (t = −1.378, p = 0.186), but CTT was significantly reduced in CGRP8–37-treated mice at D1 (t = −2.659, p = 0.013), D7 (t = −3.522, p = 0.002), and D28 (t = −2.048, p = 0.028) post-SCI.
When evaluating pellet water weight loss as a quantitative measurement of constipation, both treatment groups showed alterations in fecal water loss over time. This effect was particularly clear in the acute timepoints and was unchanged by CGRP8–37 treatment (Fig. 1D). A 2 (treatment) × 4 (time) mixed design repeated measures ANOVA confirmed a significant main effect of time (F[3,51] = 12.976, p < 0.001) on pellet weight, but no other significant main effects or interactions (all F < 1.533, all p >0.05). While intrarectal CGRP8–37 does not significantly affect recovery of locomotor function, a single dose of CGRP8–37 is sufficient to prevent dysmotility after injury, suggesting a role for CGRP in NB that is, at least partially, distinct from biological processes involved in locomotor dysfunction and recovery.
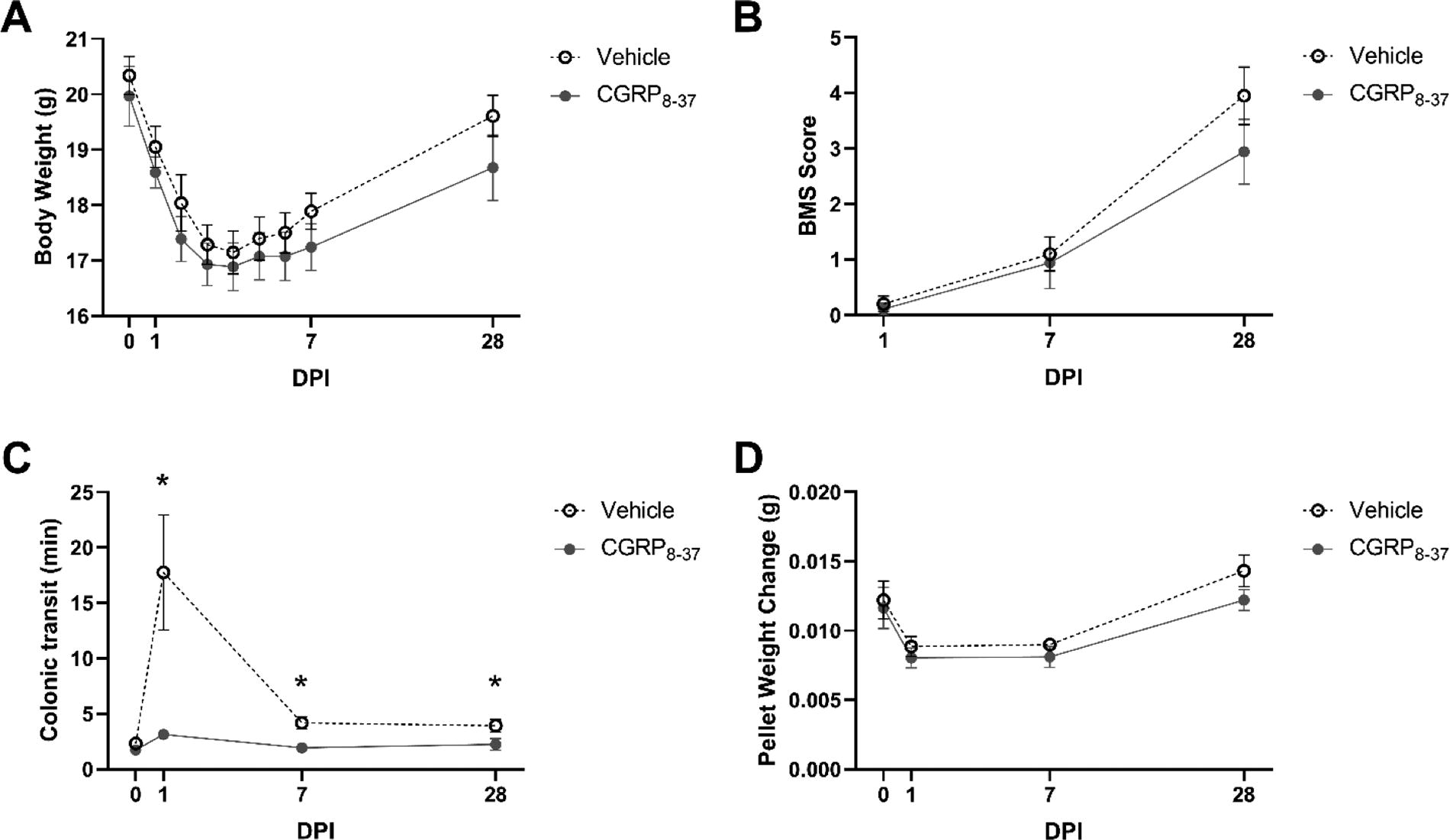
CGRP8–37 prevents acute cellular DNA damage within the Colon
We recently reported accumulation of DNA damage within cells of the colon after SCI. 20 DNA damage is produced by inflammation through free radicals and leads to cellular and/or organ dysfunction as well as impaired recovery of function after injury. 20 Thus, we tested the hypothesis that CGRP-initiated neurogenic inflammation instigates colonic cellular damage that could be prevented by administration of CGRP8–37 as measured using the comet assay as a quantitative measure of DNA damage within individual cells. Figure 2 illustrates differences between spinally injured mice treated with vehicle (panels A, B, C) or CGRP8–37 (panels A′, B′, C′) at D1, D7, and D21 post-SCI, respectively. A 2 (treatment) × 3 (time) ANOVA confirmed significant main effects of condition (F[1,354] = 12.927, p = 0.000) and treatment (F[1,354] = 12.500, p = 0.000) as well as a significant interaction of condition × treatment (F[2,354] = 5.180, p = 0.006). Independent samples t-tests at each time point revealed shorter comet tail scores for the CGRP8–37 group compared to the vehicle at both D1 (t = −4.093, p = 0.000) and D7 (t = −3.541, p = 0.000), suggesting that CGRP8–37 prevents acute cellular damage to the colon.
Single-cell gel electrophoresis (comet assay) quantification of double-stranded DNA breaks in the colon after injury. Representative images of D1 SCI
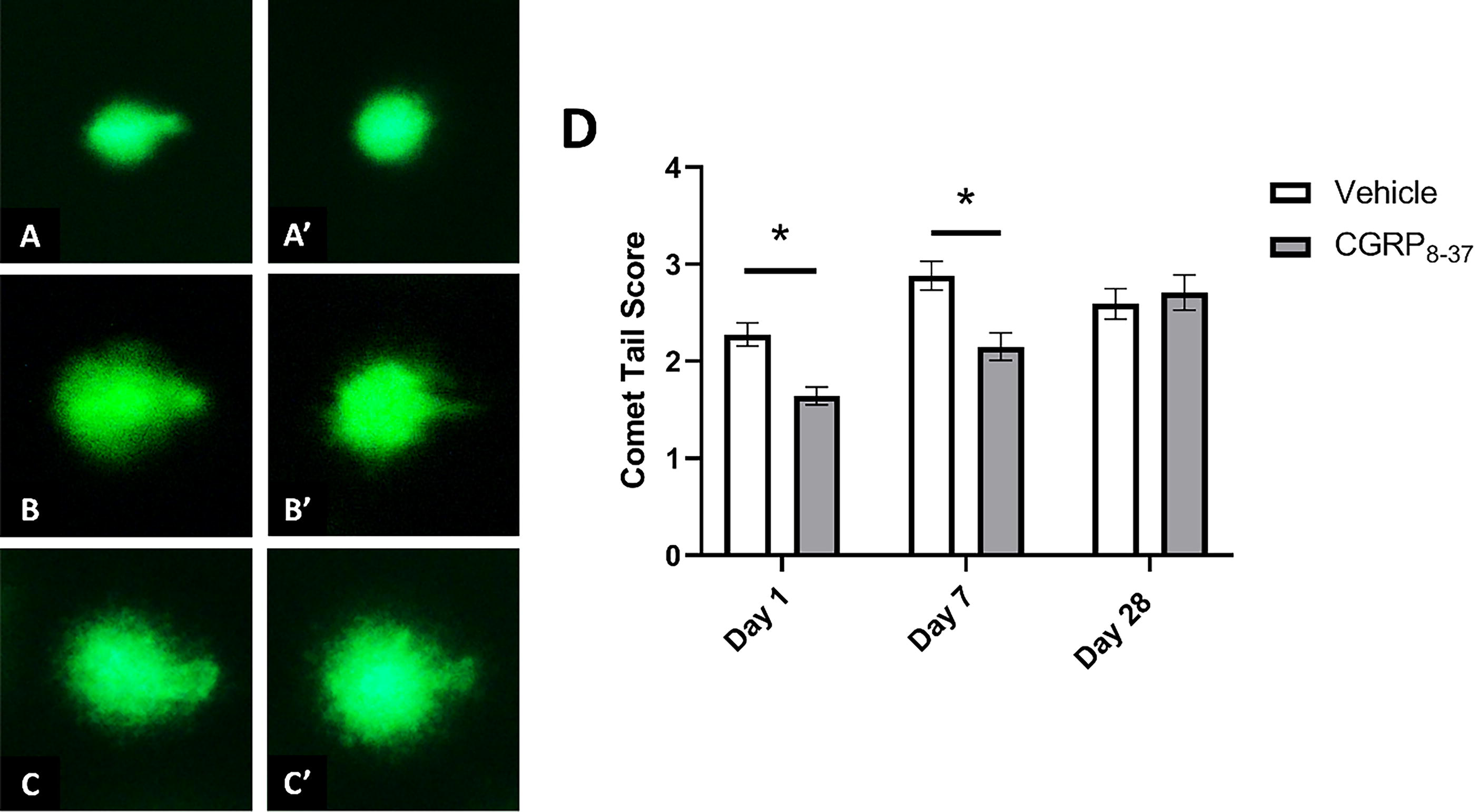
CGRP8–37 prevents acute structural defects of the colon and microbial translocation into the colon wall
Alcian blue staining of colon cross-sections was used to assess colon epithelium structure (Fig. 3). Figure 3 illustrates differences between spinally injured mice treated with vehicle (panels A, B, C) or CGRP8–37 (panels A′, B′, C′) at D1, D7, and D21 post-SCI, respectively. A 2 (treatment) × 3 (time) ANOVA revealed a significant main effect of condition (F[2,24] = 4.877, p = 0.017) as well as a significant condition × treatment interaction (F[2,24] = 4.269, p = 0.026). No other significant effects were present. Follow-up analysis using independent samples t-tests to assess group differences (vehicle vs. CGRP8–37) at individual timepoints revealed a significant increase in alcian staining only for the D1 CGRP8–37 group compared to the D1 vehicle control (t = −2.159, p = 0.045), while comparison of data from D7 post-SCI approached significance (p = 0.095).

Goblet cells are stained with Alcian blue, and sections were counterstained with Nuclear Fast Red. Representative images of D1 SCI
Given the preservation of goblet cell numbers following CGRP antagonism, we expected a subsequent improvement in the mucus layers and reduced microbial translocation out of the intestinal lumen. We used a TexasRed-conjugated EUB338 probe in FISH to quantify the sterile distance between the microbes and colon wall (Fig. 4). Figure 4 illustrates differences between spinally injured mice treated with vehicle (panels A, B, C) or CGRP8–37 (panels A′, B′, C′) at D1, D7, and D21 post-SCI. The sterile distance is defined as the blank space on the luminal side of the mucosal epithelial layer devoid of bacterial staining with the EUB338 probe. A thinner or nonexistent (i.e., bacterial invasion of host tissue) sterile distance between the colon wall and microbes suggests increased likelihood of potentially harmful host–microbiome interactions. Using the EUB338 probe to localize gut bacteria, a 2 (treatment) × 3 (time) ANOVA revealed significant main effects of condition (F[1,344] = 60.756, p = 0.000) and treatment (F[1,344] = 88.240, p = 0.000), as well as a significant treatment × time interaction (F[2,344] = 18.510, p = 0.000). Post hoc comparisons using independent samples t-tests confirmed significantly increased distance for the CGRP8–37 treatment compared to vehicle at D1 (t = 9.264, p = 0.000), D7 (t = 3.209, p = 0.001), and D28 (t = 5.390, p = 0.000), suggesting that microbial translocation is downstream of CGRP expression in this model.
FISH stain was conducted using the DNA oligo probe, EUB338. Sections are counterstained with DAPI. Representative images of D1 SCI
CGRP8–37 prevents SCI-induced vagal and enteric neuron hyperresponsiveness
We have recently published the Ca2+ influx of in vitro cultured intrinsic ENS neurons and extrinsic primary afferents innervating the colon as a proxy measure of pain pathway activation, particularly given the difficulty in behavioral assessment of pain hypersensitivity following SCI. Our previous findings indicate enhanced Ca2+ responses for colon-specific ND and ENS neurons (but not DRG), and we propose that this increased cellular response could reflect sensory dysfunction capable of supporting chronic bowel pain after SCI. 20 After SCI, these neuronal subpopulations exhibited a heightened peak Ca2+ response (ΔF) to high K+, and a higher proportion of neurons responded to fecal supernatants. 20 Not only is CGRP itself known to play a role in pain processing, but antagonism of CGRP successfully prevented microbial invasion of the colon wall in our study, suggesting the potential for microbial mediation of NB phenotypes, including abdominal pain and dysfunction. We, therefore, tested whether CGRP8–37 at the time of injury could prevent general hyperresponsiveness to high K+ as well as the specific Ca2+ response to fecal supernatants in the same neuronal populations previously assessed (Fig. 5).
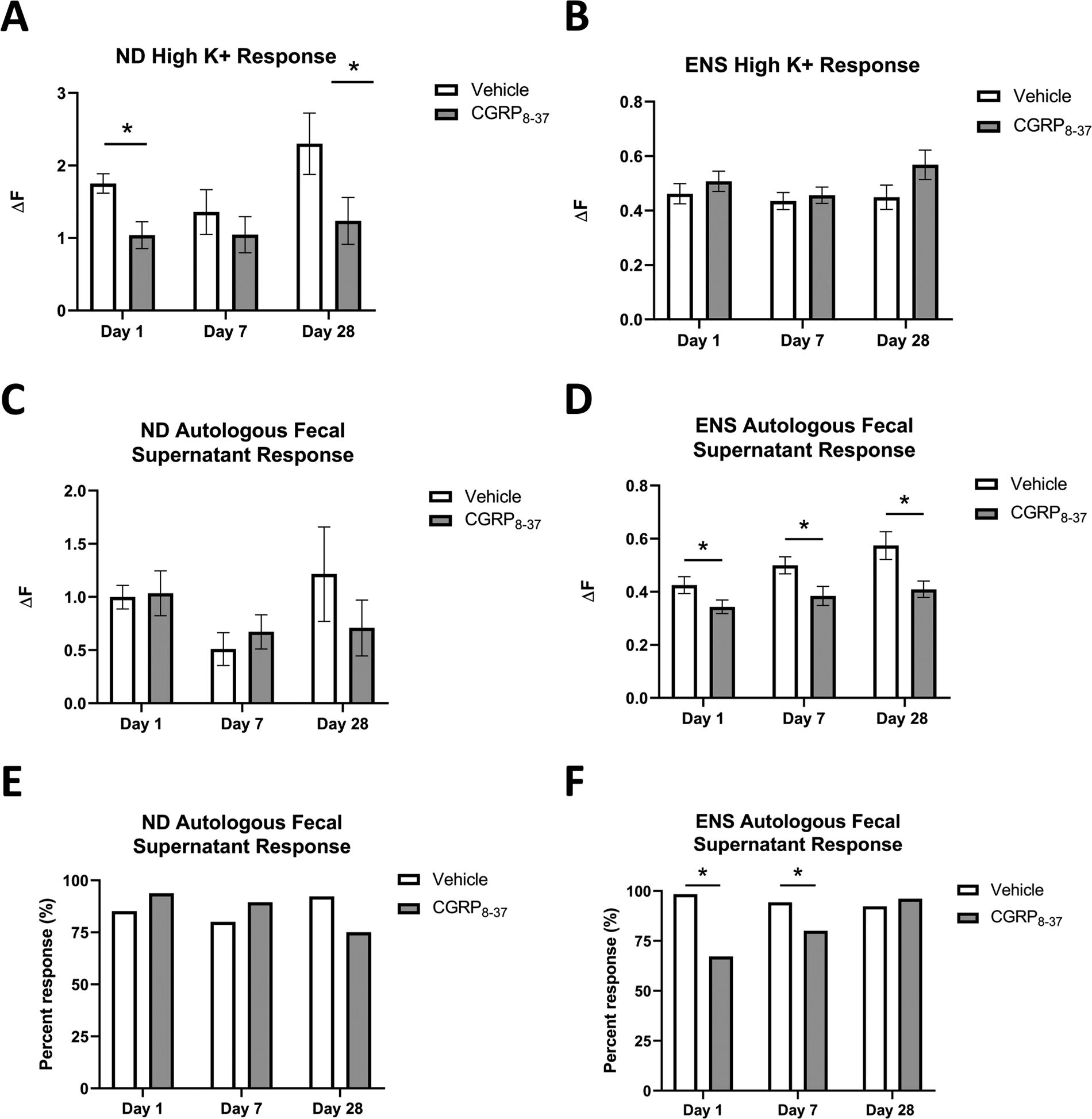
Ca2+ imaging of colon-specific, ND
The sample size in the present study reflects our prior work 20 reflecting that this sample size (2 cover-slips per cellular population/mouse, 3 mice/condition) is appropriate for the identification of differences in Ca2+ response magnitude and in the proportion of cells that respond to a given agonist. All neurons were exposed to 30 mM (high) K+ to determine viability and record the ΔF in response to forced depolarization. ND and ENS cell populations were analyzed separately. A 2 (treatment) × 3 (time) ANOVA conducted on the ΔF response in ND afferents to high K+ revealed a significant main effect of treatment (F[1,157] = 8.485, p = 0.004) but no other main effects or interactions (Fig. 5A). Post hoc independent samples t-tests confirmed significantly reduced Ca2+ transients in response to depolarization with high K+ for the CGRP8–37 groups compared to vehicle at both D1 (t = −3.133, p = 0.002) and D28 (t = −1.970, p = 0.031). We also assessed the response of ENS neurons to high K+, and there were no significant main effects or interactions with a two-way ANOVA (all F < 3.733, all p > 0.05; Fig. 5B).
We next assessed the Ca2+ influx of ND and ENS neurons in response to autologous fecal supernatants as described previously. 20 In line with our prior report, a 2 (treatment) × 3 (time) ANOVA conducted on the ΔF in ND afferents revealed no significant main effects or interactions (all F < 1.919, all p > 0.05; Fig. 5C). However, a 2 (treatment) × 3 (time) ANOVA on the ΔF of ENS neurons exposed to autologous fecal supernatants revealed significant main effects of condition (F[2,710] = 4.408, p = 0.013) and treatment (F[1,710] = 16.818, p < 0.001), but no significant interaction (F[2,710] = 0.655, p = 0.520) on the ENS Ca2+ response to autologous fecal supernatants (Fig. 5D). Post hoc independent samples t-tests confirmed reduced responses for the CGRP8–37 group compared to vehicle at D1 (t = −1.998, p = 0.047), D7 (t = −2.394, p = 0.017), and D28 (t = −2.709, p = 0.007). χ2-square tests conducted on the proportion of ND cells responding to autologous fecal supernatants indicated no difference between conditions or treatments (all χ2 < 0.754, all p > 0.05; Fig. 5E). In contrast, χ2-square tests conducted on the proportion of ENS cells responding to autologous fecal supernatants indicated a significantly higher proportion of responsive cells over time (χ2 = 24.285, p < 0.001) but a reduced proportion of ENS cells that responded in the CGRP8–37 treatment group compared to the vehicle (χ2 = 48.280, p < 0.001; Fig. 5F). We conducted a chi-squared test between vehicle and CGRP8–37 at each timepoint as a follow-up post hoc test and confirmed a reduction in proportion of cell responding at D1 (χ2 = 44.161, p = 0.001) and D7 (χ2 = 13.026, p = 0.001), but not D28 (χ2 = 1.511, p = 0.219).
Discussion
Chronic abdominal pain and colonic dysmotility are hallmark NB symptoms representing a significant burden for PWSCI. 13 However, little is known about the mechanisms underlying NB initiation and maintenance, resulting in limited therapeutic options. SCI offers a unique opportunity to study the early pathological processes that contribute to NB because neurological injury occurs suddenly, and care is sought immediately, even in the clinical setting. 20 Our recent characterization of rapid onset CGRP overexpression in the colon, in tandem with prior work implicating CGRP release from spinal primary afferents into hind paw skin after SCI, 19 pointed to a role for CGRP-mediated neurogenic inflammation in the initiation of SCI-induced NB. The current experiments support this hypothesis as intrarectal CGRP antagonism with the preclinical peptide CGRP8–37 prevents NB phenotypes (i.e., colonic dysmotility, neuronal hyperresponsiveness) as well as other biological processes (i.e., genomic instability, goblet cell decrease, microbial invasion).
Reports from our lab and others clearly indicate that mice with SCI have a limited behavioral repertoire reflective of deficits seen clinically. The inability to generate an organized visceromotor response to colorectal distension, however, does not necessarily indicate normal pain sensitivity or hypoalgesia. To shed light on the mechanisms of NB pain without depending on behavior, we employ Ca+2 imaging to examine responses in extrinsic and intrinsic neurons with a potential role in pain signal transmission. Our findings suggest that intrarectal CGRP antagonist treatment at the time of spinal injury may attenuate enteric and vagal neuronal hyperresponsiveness. The prevention of hyperresponsiveness in colon-specific vagal sensory neurons is particularly promising, as these neurons provide a direct path for pain signals to bypass the injured spinal cord but still reach the brain.
CGRP has already been implicated in migraine pathogenesis as well as in the rarer painful condition of abdominal migraine 39 where the hallmark symptoms are gastrointestinal dysfunction and visceral pain of unknown origin, 39 similar to the symptom profile of NB. While a number of FDA-approved CGRP antagonists are recommended for treating migraine, these antagonists have not been directly tested for the treatment of abdominal migraine. Interestingly, anti-CGRP molecules have been shown to alleviate both heightened sensitivity and organ cross-sensitization in animal models of visceral hypersensitivity,40,41 and anti-CGRP medications have been proposed for the treatment of constipation by restoring peristalsis, given that CGRP is known to inhibit colon muscle contractions,42–46 suggesting that intrarectal antagonism of CGRP could be a beneficial application to multiple conditions characterized by altered colonic transit and/or abdominal pain dependent on CGRP overexpression.
We further propose that CGRP plays a role in intestinal barrier function, potentially through the initiation of localized inflammation, instead of the plausible alternative hypothesis that microbial invasion initiates CGRP overexpression as a mechanism of the host’s defense. Should the alternative hypothesis be correct, we would have expected CGRP antagonism to worsen microbial outcomes. The observed prevention of microbial translocation into the colon wall and prevention of neuronal hyperresponsiveness to fecal supernatants is exciting because it (1) supports the role of CGRP in the regulation of intestinal barrier integrity and (2) points to the therapeutic potential of CGRP antagonist treatment in SCI patients to prevent initiation of pathophysiological processes resulting in NB. Moreover, maintaining a functional intestinal barrier and restricting bacteria to the intestinal lumen could reduce the risk of septicemia, one of the most common reasons for SCI patients to seek postrehabilitative emergency medical treatment.47–50
Limitations and future directions
CGRP8–37 is a preclinical peptide with a short efficacy window of less than 30 min in rodents, 51 while the CGRP overexpression window persists in our model for at least 24 h. Our data suggest that even brief CGRP antagonism at the time of injury may be enough to disrupt the pathological processes underlying NB at the acute time points, with long-term prevention of select outcomes, but future studies could assess longer-term CGRP targeting by leveraging currently available FDA-approved CGRP monoclonal antibodies used as a monthly preventative for migraine.52,53 An exciting future direction for this study would be to administer one of these longer-acting medications in mouse models of SCI with the goal of improving clinical translatability by preventing CGRP overexpression across the entire acute SCI period and increasing viability of therapeutic intervention.
We are the first, to our knowledge, to use intrarectal administration for this specific antagonist. While the goal of this approach was to leverage a well-characterized molecule for a new application, it leads to ambiguity in the mechanism of action. CGRP8–37 has primarily been used intrathecally, which offers greater mechanistic clarity due to the relative cell type homogeneity represented in the dorsal root ganglia, for example. On the contrary, the cellular makeup of the colon is vastly more heterogeneous and is known to utilize protective barriers for selective permeation into the colon wall. While our results are promising, it is unclear whether CGRP8–37 crosses the intestinal mucosal layer(s) or if it remains confined to the lumen. This lack of clarity as to whether the antagonist is acting on luminal-facing cells or on cell types within the colon wall or nerve endings of extrinsic afferents innervating the gut means that we do not know the cell type that CGRP8–37 acts on to prevent NB phenotypes. Future studies are needed to disentangle this issue.
Conclusions
Most spinally injured patients immediately seek emergency medical care, meaning that patients are uniquely positioned to receive preventative therapeutics designed to prevent rather than treat long-term complications. However, the mechanisms of chronic gastrointestinal complications after SCI remain poorly understood. To begin filling the knowledge gap around mechanisms of NB after SCI, we used a preclinical model of SCI-induced NB and found that intrarectal CGRP antagonism at the time of SCI prevents colonic dysmotility, structural damage to the colon, and neuronal hyperresponsiveness. This supports our hypothesis that CGRP release initiates NB phenotypes, including neuronal hyperresponsiveness due to host–microbiome interactions downstream of colonic homeostasis defects. Continued investigation into the cause-and-effect relationship of CGRP in NB will expand our basic understanding of NB pathogenesis and, hopefully, advance the development of novel therapeutic strategies for the treatment or prevention of NB at the time of injury.
Transparency, Rigor, and Reproducibility Summary Statement
A sample size of 10 mice per group was planned for assessments of overall health and behavioral assays (i.e., CTT and fecal pellet water weight). Molecular and histological experiments were planned for five mice per group, while calcium imaging experiments were planned for three mice per group (80–100 cells per mouse for ENS and 2–5 per mouse cells for nodose ganglia, given the reduced colon-specific cellular representation in the vagus). While the differences in the number of cells included from the two cellular subpopulations are substantial, the ENS and ND cellular responses were never compared to each other, and all analyses were conducted within a cellular subpopulation, and the total number of cells used from each condition was not significantly different. Actual sample sizes for all experiments remained as planned except for one loss in the treatment group for the behavioral assays, leading to a sample size of n = 9. There is otherwise no missing data since potential outliers were not excluded from the dataset. Eight- to ten-week-old mice were ordered and shipped to the University of Kansas Medical Center and allowed to acclimate in the animal facility for two days before surgery. This established age- and shipment-matched subjects to increase rigor. However, this study was restricted to female mice because bladder voiding efficacy is low in males, leading to reduced survival as discussed in our recent publication. 20 Mice were randomly selected from a cage and sorted into three different timepoints postinjury, as well as either vehicle or CGRP8–37 treatment. Behavioral analyses were not blinded to investigators, but histological and cellular experiments were blinded and scored by two independent researchers. All equipment and reagents used to perform this study are listed after the first instance mentioned in the document and are available for purchase. The authors agree to publish the article under a Creative Commons Open Access license, and the data will be made available from the authors upon request.
Authors’ Contributions
A.B.W.: Conceptualization, methodology, validation, formal analysis, investigation, supervision, and funding acquisition. L.K., S.C., M.E., and S.M.: Investigation, formal analysis, and visualization. J.C. and K.B.: Methodology, resources, and supervision. E.Y.: Conceptualization, methodology, validation, formal analysis, investigation, resources, supervision, and funding acquisition. All authors approved the final article.
Footnotes
Author Disclosure Statement
The authors declare that they have no competing financial interests or personal relationships that could have influenced the work reported in this article.
Funding Information
This work was supported by NIH grants R03 NS096454 (K.M.B.) and R21 NS104789 (K.B.), the Rita Allen Foundation Award in Pain (K.B.), the Kansas Institutional Development Award (IDeA) P20 GM103418 (E.Y.), Craig H. Neilsen SCIRTS grant (E.Y.), the KUMC Biomedical Research Training Program (A.B.W.), the Kansas University Training Program in Neurological and Rehabilitation Sciences supported by NIH award number T32 HD057850 (A.B.W.), and core support from the Kansas IDDRC P30 HD000228.
Data Availability
Data will be made available by request.