
Abstract
Background
Despite advances in therapeutics, hepatocellular carcinoma (HCC) remains one of the most malignant types of digestive tract cancers with a poor prognosis. Pyroptosis is a form of programmed cell death induced by inflammatory caspases. Recent studies have identified pyroptosis, a form of programmed cell death induced by inflammatory caspases, as playing a role in tumorigenesis and cancer progression. However, the functions and mechanisms of pyroptosis in HCC are barely explored.
Methods
Gene expression and clinical data were derived from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. A prognostic signature and nomogram were constructed by on differentially expressed genes and clinical data. Pathway enrichment and immune cell infiltration were further analyzed. Potential drugs to modulate the pathways were explored.
Results
In this study, a pyroptosis-related gene signature was developed and identified to be significantly correlated with the survival of HCC patients. Additionally, a nomogram on the basis of pyroptosis-related genes was constructed with distinct prognostic values. Furthermore, the pyroptosis-related gene signature might correlate with immune-related pathways and the regulation of the immune microenvironment, and several compounds (KIN001-220, TPCA-1, LY-303511, physostigmine, vemurafenib, etc.) could potentially reverse the pathogenic gene-expression patterns.
Introduction
Liver cancer remains the fourth most common cause of cancer-related mortality. 1 Hepatocellular carcinoma (HCC) is the predominant type, accounting for over 90% of all primary liver cancer cases. 2 Despite rapid advancements in treatments such as surgery, radiotherapy, chemotherapy, targeted therapy, and immunotherapy/immune checkpoint inhibitors, 3 the overall five-year survival rate for liver cancer remains low at approximately 12%. Therefore, identifying, novel biomarkers or gene signatures could significantly aid in both the diagnosis and prognosis prediction of HCC patients, as well as the development of new therapeutic approaches.
Pyroptosis is a form of programmed necrotic cell death mediated by the gasdermin family (GSDM), which act as mediators by forming pores in the plasma membrane, leading to cell swelling and lysis.4,5 Recent research has highlighted the role of pyroptosis in cancer. 6 For instance, Sorafenib, a first-line treatment for HCC, has been shown to trigger macrophage pyroptosis and enhance natural killer cell-mediated cytotoxicity in HCC therapy. 7 Moreover, GSDME-dependent pyroptosis, induced by miltirone, a phenanthrene-quinone derivative, inhibits tumor growth both in vitro and in vivo. 8 Nevertheless, a thorough evaluation of pyroptosis-related genes in HCC is still needed, as their prognostic implications and associated pathways remain largely unexplored. 9
In the present study, we conducted a comprehensive evaluation of the expression profiles, prognostic significance, enriched pathways, and correlation with the tumor immune microenvironment of pyroptosis-related genes in HCC. Furthermore, we developed a novel pyroptosis-gene-based signature for predicting HCC prognosis and explored potential drugs to provide new insights into targeted therapy.
Methods
Training and validation datasets
We obtained the RNA-sequencing (RNA-seq) data of 424 patients and clinical information from The Cancer Genome Atlas (TCGA) database. In addition, the validation dataset was acquired from the Gene Expression Omnibus (GEO) database (GSE76427), which contained mRNA expression profiles from 115 primary tumors tissue samples and 52 adjacent non-tumor tissues samples derived from 115 HCC patients.
Identification of differentially expressed genes (DEGs)
The Gene Ontology (GO) term pyroptosis and previous review found A total of 58 pyroptosis-related genes.10–13 DEGs (P < 0.05) between the normal samples and tumor were explored using the “limma” package after normalization of the expression data. 14 The gene-interaction networks were developed by the “igraph” package. 15
Pyroptosis-related gene prognostic model development
We utilized LASSO Cox regression analysis on all identified DEGs to create a refined prognostic model.
16
Immunohistochemistry (IHC) staining images of the genes in the model were sourced from The Human Protein Atlas database.
17
The formula used for calculating the risk score was listed as follows: risk score =
Functional enrichment analysis
DEGs were identified with criteria of |log2FC| ≥ 1 and a false discovery rate (FDR) < 0.05. Based on the “clusterProfiler” package, we performed the pathway enrichment for Kyoto Encyclopedia of Genes and Genomes (KEGG) terms and GO.21–24
Tumor microenvironment analysis
We analyzed the correlation between pyroptosis-related DEGs and immune infiltration, using the Tumor Immune Estimation Resource (TIMER) database. 25 The TIMER database also enabled the comparison of immune scores among subtypes. To investigate differences in the composition of 22 immune cell types between low- and high-risk subgroups, the CIBERSORT algorithm was applied. 26 Single sample gene set enrichment analysis (ssGSEA) was carried out with the “gsva” package, utilizing 29 immune gene sets to calculate infiltrating scores for immune cells in each sample and to evaluate immune-related pathways. 27
Exploration of potential compounds
Connectivity map (CMap) (https://clue.io/) 28 was used to identify potential compounds based on the top 150 genes. The induction effect of the compound to the signature is represented by positive scores, whereas the inhibition effect is represented by negative scores. The larger absolute values of the scores indicate stronger effects.
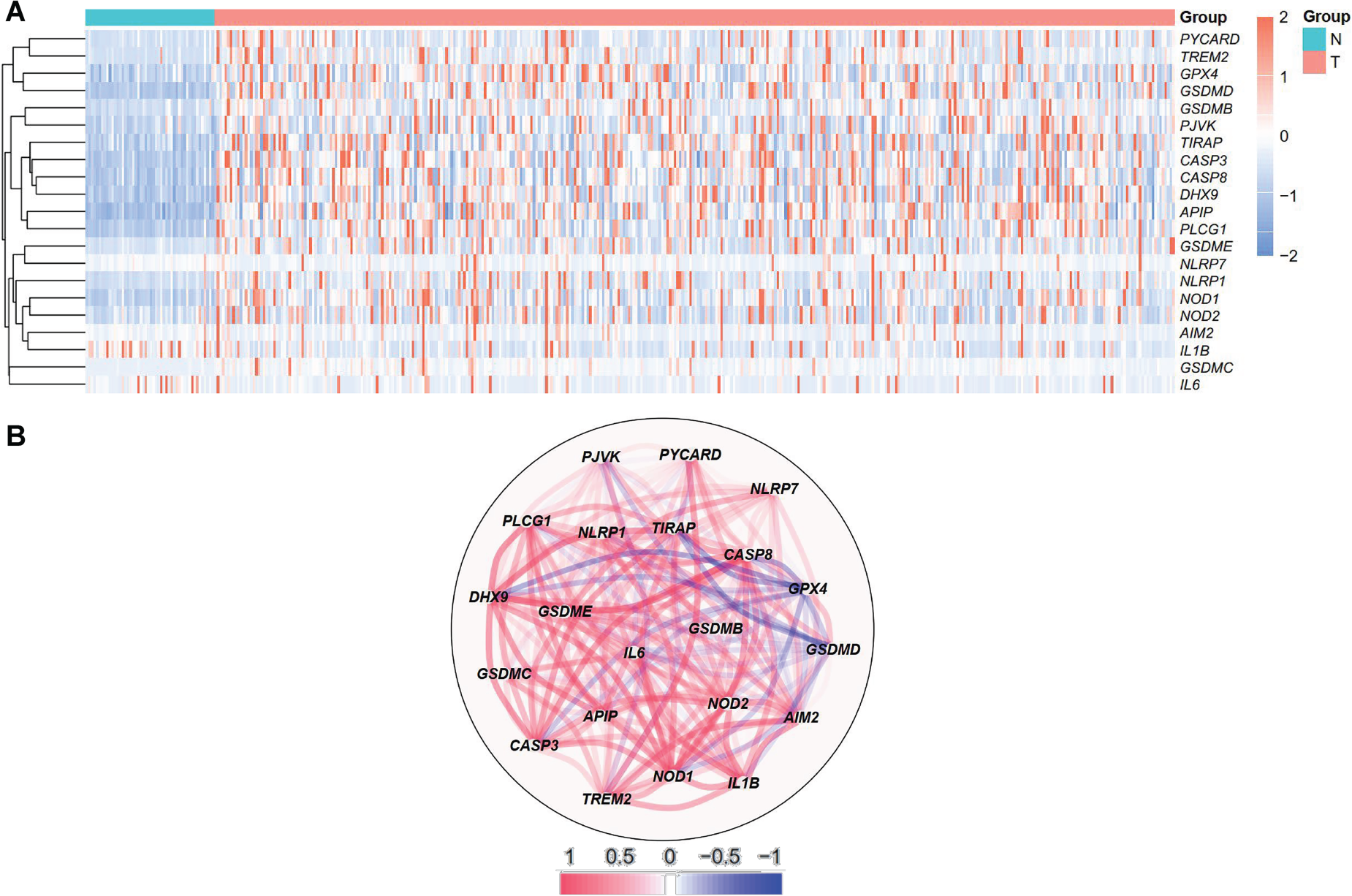
Expression profile of 21 differentially expressed pyroptosis-related genes in HCC. A. Heatmap demonstrating gene expression in the tumor (T) and normal tissue (N) group. B. Correlation network of 21 differentially expressed genes.
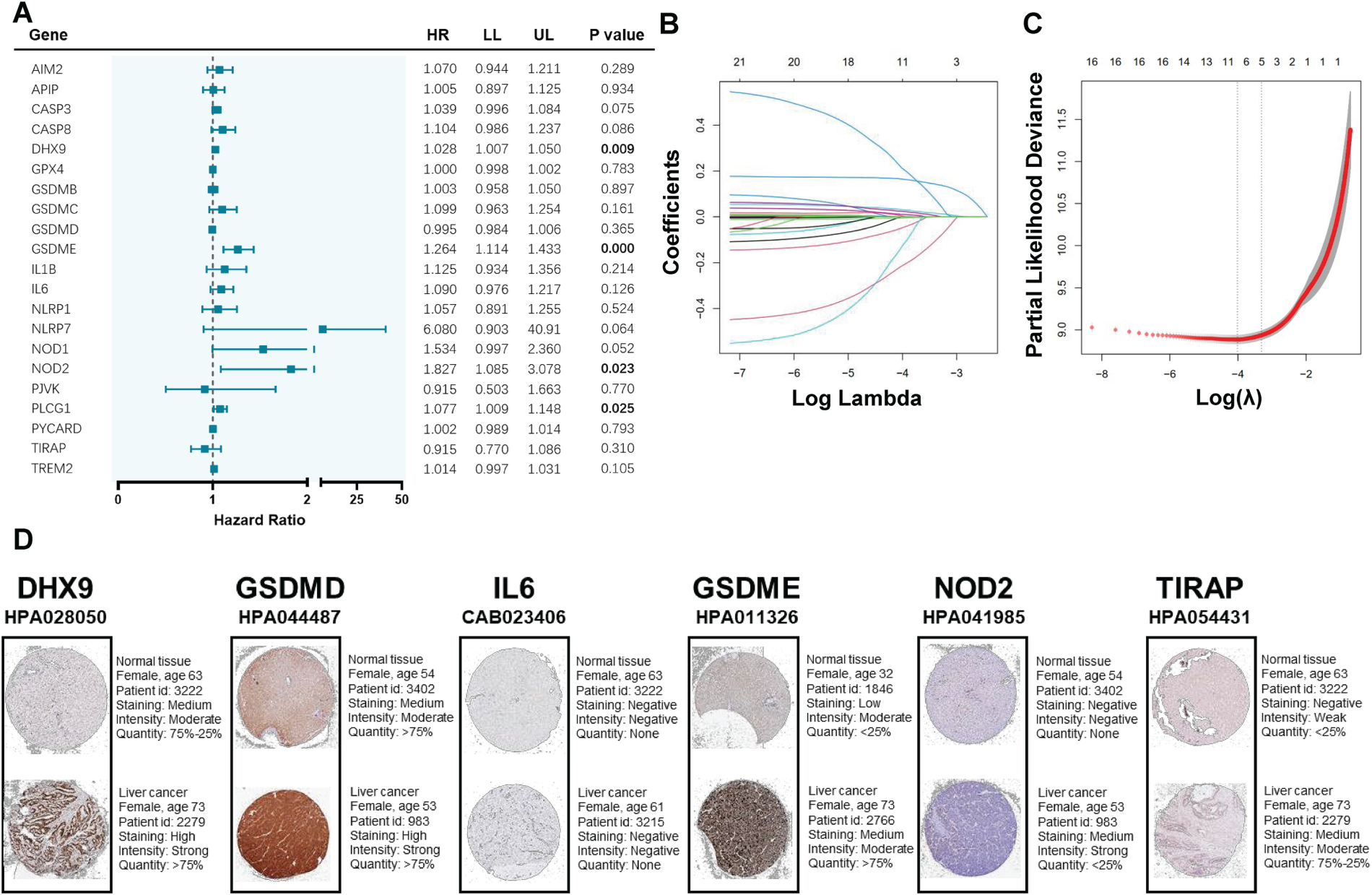
Construction of a pyroptosis-related gene signature in the TCGA cohort. A. Univariate cox regression analysis of overall survival (OS) for the selected genes. B. LASSO cox regression of the 21 selected genes. C. Cross-validation in lasso regression. D. Immunohistochemistry staining of selected genes in HCC and normal liver tissues.
Statistical analyses were executed using R software (version 4.1.0) and SPSS software (version 25.0). Continuous variables were analyzed using the t-test or the Wilcoxon rank sum test, depending on their normality. The Pearson chi-square test was applied for differences in categorical variables. The Kaplan-Meier and log-rank test method were utilized to evaluate survival between subgroups. Multivariate Cox regression analysis identified the independent prognostic factors.
Results
Identification of DEGs in HCC
Analyzing the TCGA data, the expressions of 58 pyroptosis-related genes were assessed, identifying 21 DEGs with |log2FC| ≥ 0.5 and P < 0.05. Among the DEGs, GSDMC, TREM2, PLCG1, PYCARD, GSDMB, GSDME, NLRP1, GSDMD, APIP, CASP8, NOD1, DHX9, CASP3, PJVK, TIRAP, NOD2, GPX4, NLRP7 and AIM2 were more expressed in the HCC group, while the expressions of IL1B and IL6 were downregulated compared to those in the normal tissues (Figure 1A). The correlation network containing the DEGs was demonstrated in Figure 1B, and strong correlations of expression levels were observed among them.
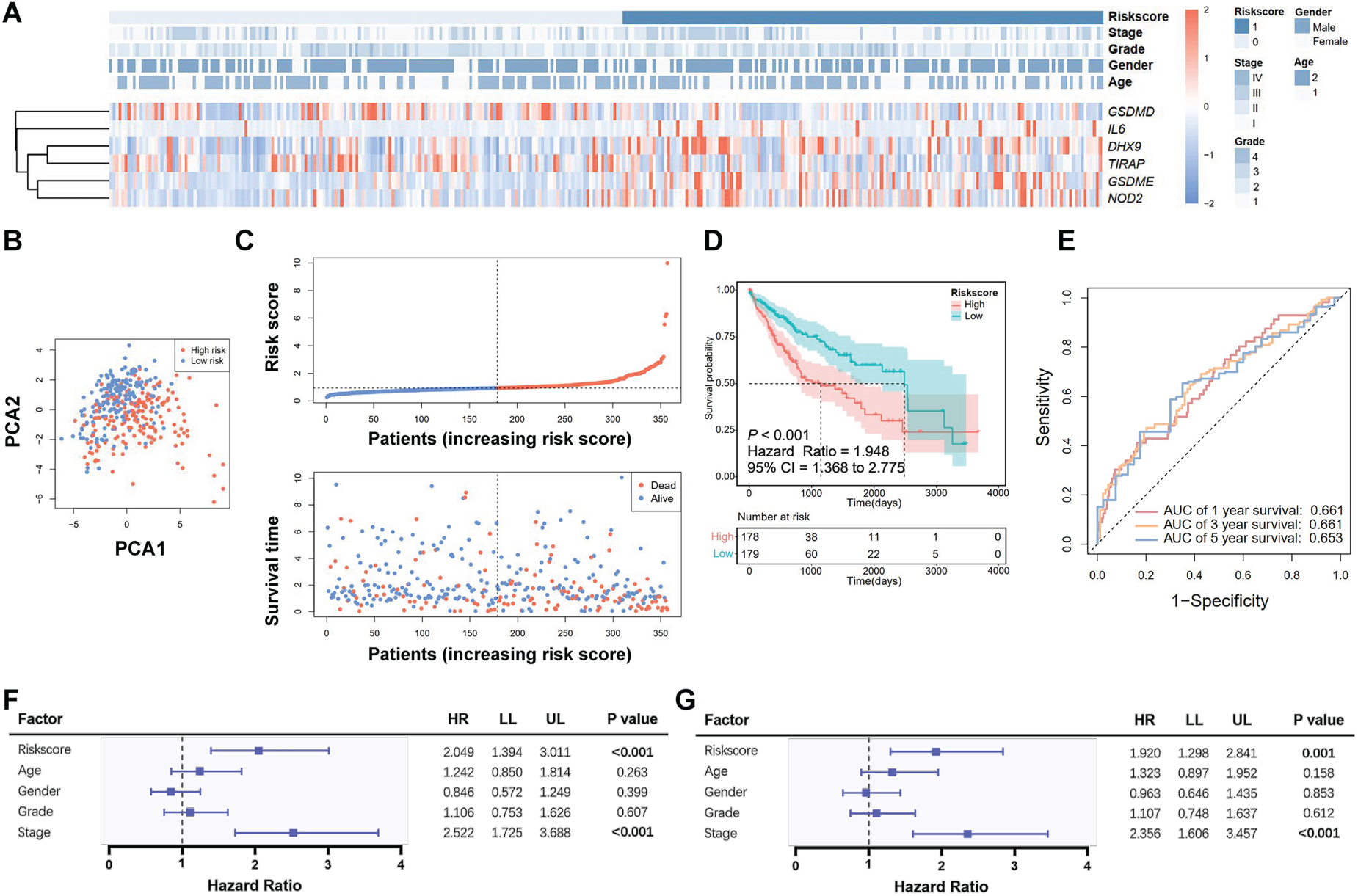
Prognosis prediction by the pyroptosis-related gene signature in TCGA cohort. A. Heatmap of the selected gene expressions with clinical features. B. Principal component analysis (PCA) of the risk groups. C. Distribution of risk scores and survival time. D. Kaplan-Meier curves for the survival of patients. E. Receiver operating characteristic (ROC) curve for 1-, 3- and 5-year survival. F. Univariate Cox analysis for predicting factors. G. Multivariate Cox analysis for predicting factors.
To develop a pyroptosis-related prognostic gene model, 357 HCC patients with available survival data were included. Figure 2A demonstrates the prognostic values of DEGs by Cox regression analysis. LASSO Cox regression analysis was conducted and a total of 6 genes (DHX9, GSDMD, GSDME, IL6, NOD2 and TIRAP) were retrieved in the prognostic model (Figure 2B, 2C). The risk score was calculated as (0.017 * DHX9) + (−0.001 * GSDMD) + (0.146 * GSDME) + (0.008 * IL6) + (0.098 * NOD2) + (−0.123 * TIRAP). In the Human Protein Atlas database, the expressions of DHX9, GSDME, and NOD2 were upregulated in liver cancer tissues (Figure 2D).
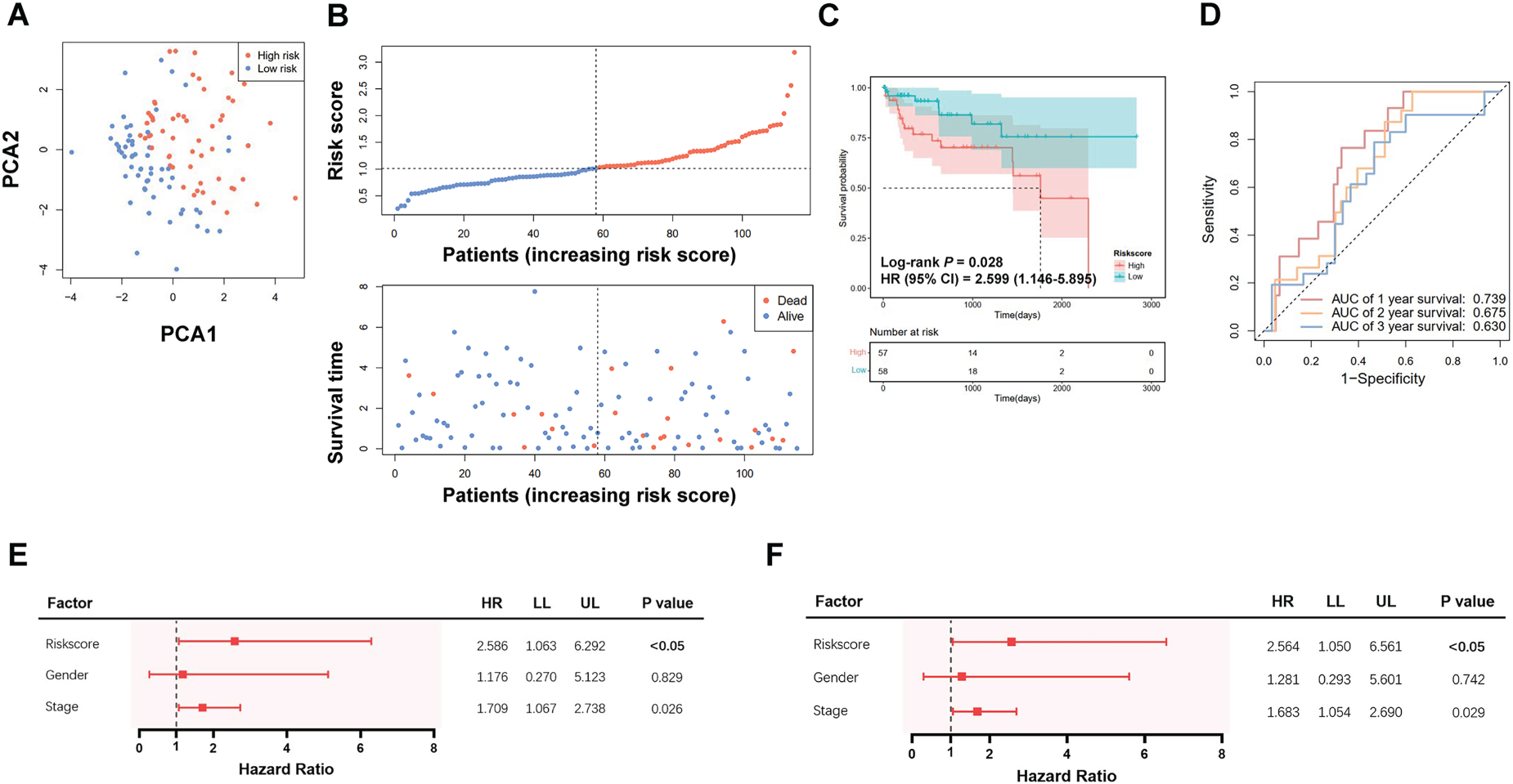
Validation of the pyroptosis-related gene signature in the GSE76427 cohort. A. Principal component analysis (PCA) of the risk groups. B. Distribution of risk scores and survival time. C. Kaplan-Meier curves for the survival of patients in the low- and high-risk groups. D. Receiver operating characteristic (ROC) curve for 1-, 2- and 3-year survival of patients. E. Univariate Cox analysis for predicting factors. F. Multivariate Cox analysis for predicting factors.
According to the risk score, HCC patients were divided into two groups, with the risk score distribution and clinical characteristics shown in Figure 3A. Figure 3A illustrates that patients with higher risk scores exhibited increased expressions of IL6, DHX9 M, GSDME, and NOD2 in cancer tissues, while GSDMD and TIRAP expressions were reduced. PCA analysis demonstrated the division of patients into two subgroups (Figure 3B). The distribution of risk scores and survival times is depicted in Figure 3C. The Kaplan-Meier curve indicated that patients with higher risk scores had poorer OS compared to those with lower scores (P < 0.001, Figure 3D), with AUCs of 0.661, 0.661, and 0.653 for 1-, 3-, and 5-year survival, respectively (Figure 3E). The risk score (low vs. high) derived from the pyroptosis-related gene signature was an independent prognostic factor for HCC patient survival (Figure 3F, 3G).
The validation dataset (GSE76427) contained Normalized expression profiles and clinical data of 115 HCC patients in the GSE76427 dataset. Because GSDME was not present in the dataset, DFNA5 was used for the prediction model. PCA analysis indicated a clear separation among HCC patients (Figure 4A), and Figure 4B presents the distribution of risk scores along with survival time. Patients with higher risk scores had notably poorer survival compared to those with lower scores (P = 0.028; Figure 4C), with AUCs of 0.739, 0.675, and 0.630 for 1-, 2-, and 3-year survival, respectively (Figure 4D). The risk score based on the pyroptosis-related gene signature proved to be an independent prognostic factor for HCC patient survival (Figure 4E, 4F).
Construction of predictive nomogram
Considering clinical features and pyroptosis-related genes, a predictive nomogram was developed based on the TNM stage, which is the standard and most commonly used staging system in clinical practice, and the risk score to predict patient prognosis (Figure 5A). The nomogram showed good calibration and added value compared to the TNM stage, the treat-all-patients approach, and the treat-none approach (Figure 5B, 5C). The AUCs of the risk score were comparable to those of the TNM stage (Figure 5D, 5E, 5F). When combined, the nomogram's AUCs for predicting 1-, 3-, and 5-year survival were 0.716, 0.752, and 0.712, respectively (Figure 5D, 5E, 5F).

Construction of a nomogram based on the pyroptosis-related gene signature. A. Nomogram for predicting the survival of HCC patients. B. Calibration curve of nomogram. C. Decision curve analysis (DCA) curve of the nomogram. D, E, F. Receiver operating characteristic (ROC) curve evaluating the efficiency of nomogram for 1-, 2- and 3-year survival of patients.
To elucidate the potential function of pyroptosis-related genes, 1631 DEGs (1612 upregulated and 19 downregulated) were identified (Supplementary Table S2). Functional enrichment analysis based on the GO database (Supplementary Table S3) demonstrated that pyroptosis-related genes were primarily involved in the regulation of humoral immune response, immunoglobulin-mediated immune response, immunoglobulin complex, B cell-mediated immunity, and channel activity (Figure 6A, 6B). KEGG pathway analysis (Supplementary Table S4) indicated that pyroptosis-related genes were mainly associated with PI3K-Akt signaling pathway, and cytokine-cytokine receptor interaction (Figure 6C, 6D).

Functional enrichment analysis of differentially regulated genes (DEGs). A, B. DEGs and pathways clustered by GO pathway enrichment analysis. C, D. DEGs and pathways clustered by KEGG pathway enrichment analysis (https://www.genome.jp/kegg/).
The analysis by CMap identified 2333 drugs with negative scores which could potentially inhibit the altered DEGs in the high-risk group. The aurora kinase inhibitor
Identification of the five most significant small molecular compounds to potentially target altered expression of differentially expressed genes (DEGs) in the high-risk group compared to the low-risk group.
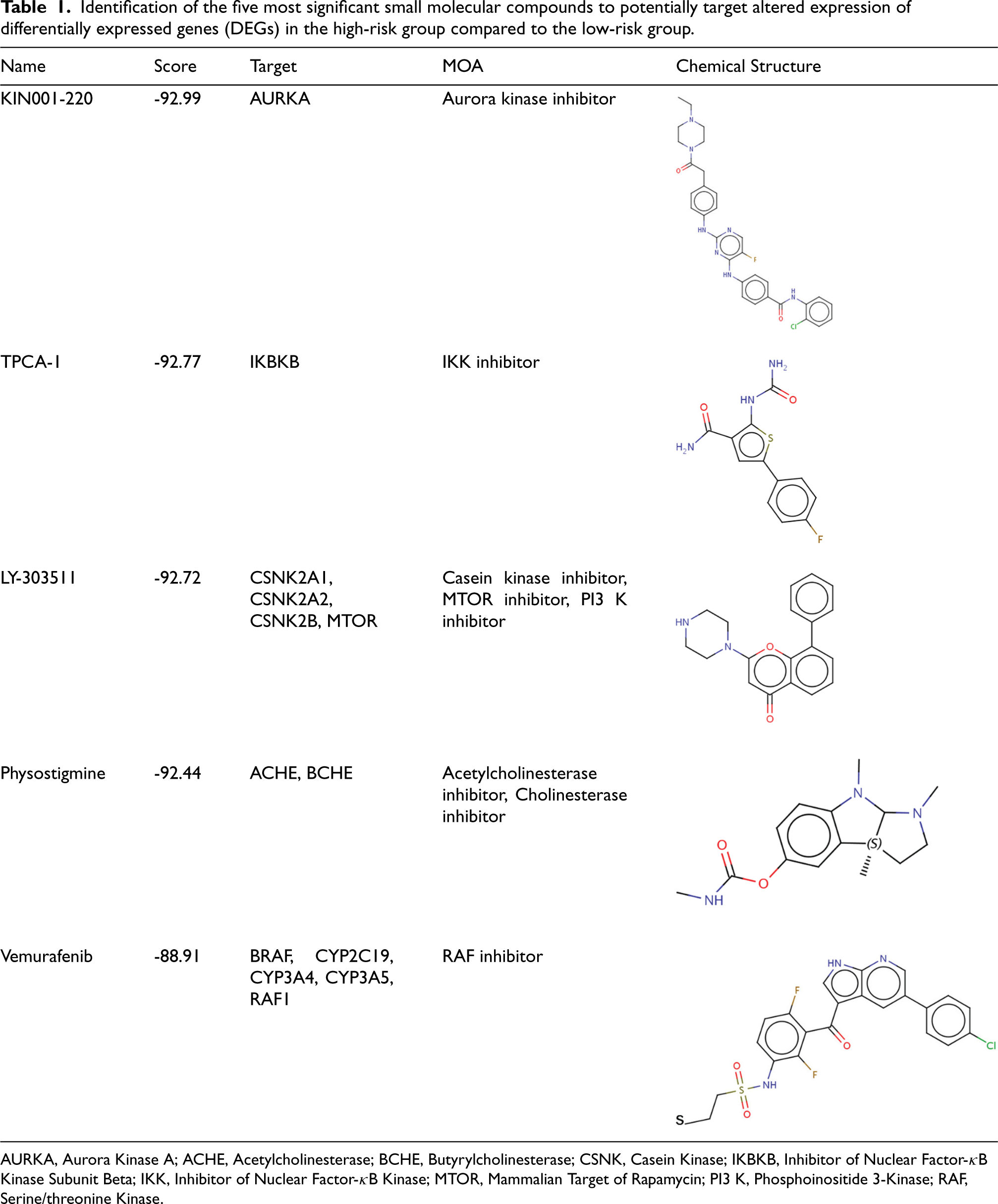
Identification of the five most significant small molecular compounds to potentially target altered expression of differentially expressed genes (DEGs) in the high-risk group compared to the low-risk group.
AURKA, Aurora Kinase A; ACHE, Acetylcholinesterase; BCHE, Butyrylcholinesterase; CSNK, Casein Kinase; IKBKB, Inhibitor of Nuclear Factor-κB Kinase Subunit Beta; IKK, Inhibitor of Nuclear Factor-κB Kinase; MTOR, Mammalian Target of Rapamycin; PI3 K, Phosphoinositide 3-Kinase; RAF, Serine/threonine Kinase.
The expressions of DHX9, IL6, NOD2, and TIRAP were highly correlated with immune cell infiltration levels, including dendritic cells, macrophages, CD8+ T cells, CD4+ T cells, neutrophils, and B cells in HCC (Figure 7A). However, there was less pronounced correlation between GSDMD expression and immune cell infiltration (Supplementary Figure S1). Somatic copy number alterations of CASP1 and GSDMB were significantly associated with immune cell infiltrations (Figure 7B, 7C), while alterations in IL6, NOD2, and TIRAP were not significant (Supplementary Figure S2). Figure 8A presented an overview of immune cell compositions. Figure 8B showed the immune cell compositions in low-risk and high-risk groups. The high-risk group had higher infiltration of B memory cells, plasma cells, activated memory CD4+ T cells, follicular helper T cells, and neutrophils, and lower infiltration of activated natural killer (NK) cells compared to the low-risk group (Figure 8C). The correlation of 22 immune cell types revealed a positive association between CD8+ T cells and activated memory CD4+ T cells (Figure 8D). In the GSE76427 cohort, immune cell infiltration levels were generally lower in the high-risk group compared to the low-risk group (Supplementary Figure S3).
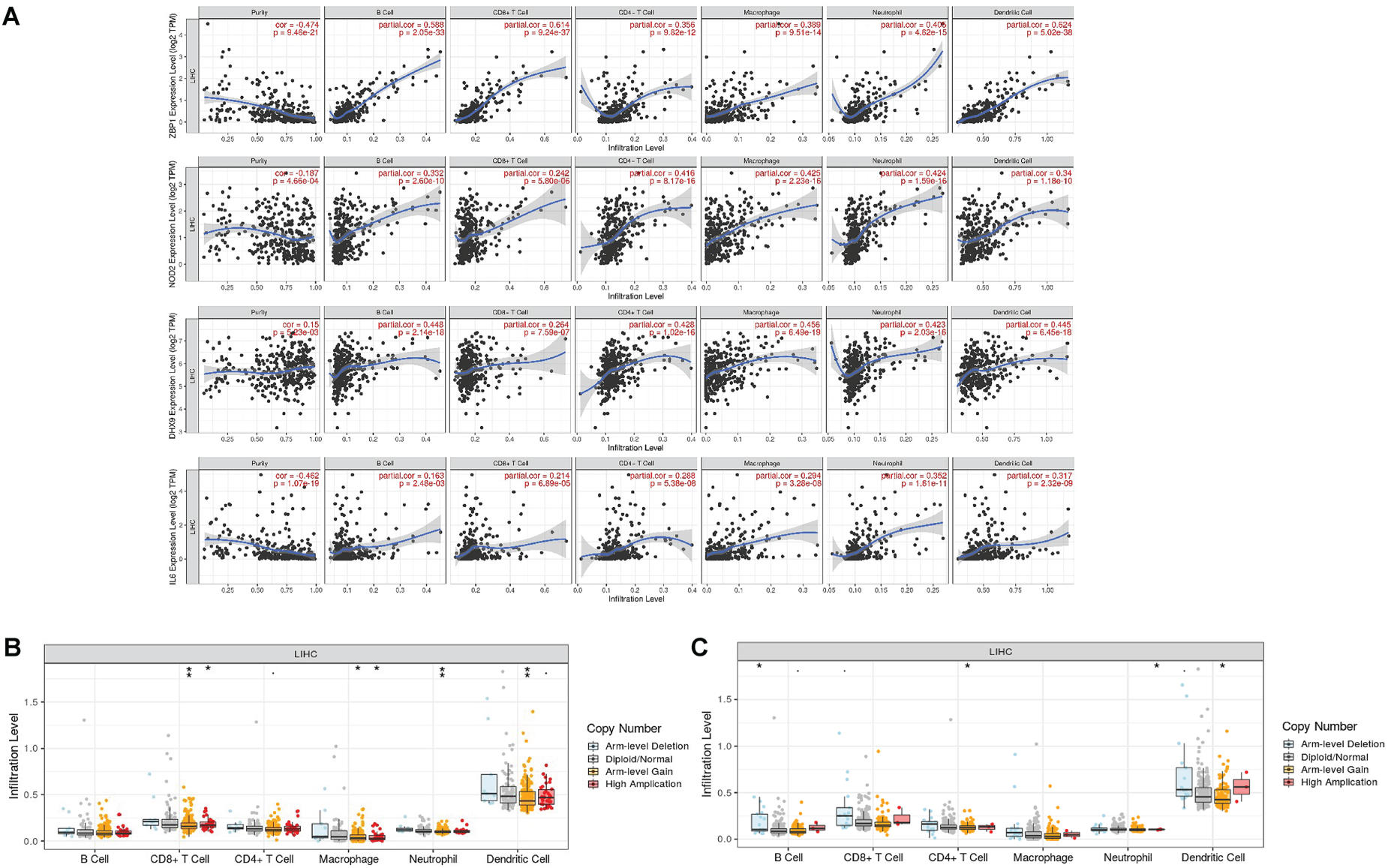
Correlations between immune cells and selected genes. A. Gene expressions (DHX9, IL6, NOD2 and ZBP1) and immune cell infiltrations in hepatocellular carcinoma. B, C. Mutants of DHX9 (B), IL6 (C) and immune cell infiltration levels in hepatocellular carcinoma.
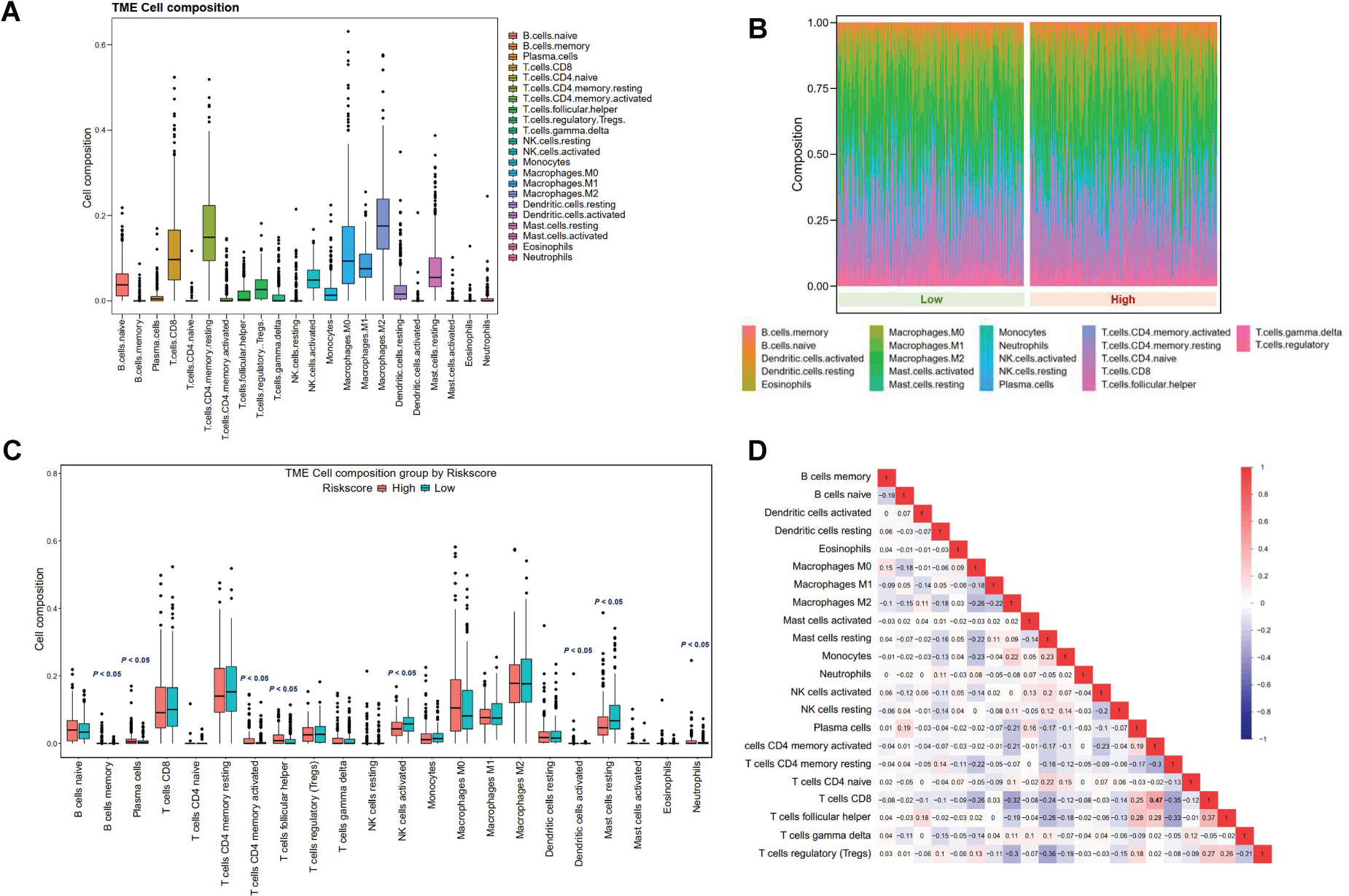
Immune cell composition in the tumor microenvironment of low- and high-risk groups. A. Overview of immune cell composition. B, C. Differences in immune cell composition classified by risk groups. D. Correlations of immune cell infiltration.
Pyroptosis is an essential, recently identified form of programmed cell death, though its role in cancer is still debated.13,29 Although evidence of pyroptosis's involvement in HCC exists, its precise function remains largely unexplored. Previous studies had developed pyroptosis-related signatures for HCC. However, deficiencies can be found for the studies, including low predictive efficacy of the signature, no construction of nomogram, no involvement of key pyroptosis-related genes, or incomprehensive assessment for tumor microenvironment.30–32 Furthermore, none of the previous studies had explored potentially therapeutic drugs that could alter the dysregulated pathways by in silico methods. Our study addresses these gaps by constructing a distinct pyroptosis-related signature, which may enhance the predictive accuracy and address the deficiencies identified in previous research.
The RNA helicase DExH-Box Helicase 9 (DHX9) is an interacting partner of NLR Family Pyrin Domain Containing 9 (NLRP9) and viral double-stranded RNA upon virus infraction, and assists in the activation of NLRP9b inflammasome to induce pyroptosis. 33 Notably, DHX9 is upregulated in various tumor tissues, including HCC, where its expression is linked to poorer patient survival.23,34 Gasdermin D and (GSDMD) and gasdermin E (GSDME) are critical members of the gasdermin family.35,36 GSDMD is the best-characterized member of the gasdermin family and considered as the pyroptosis execution protein, which is cleaved by caspases and induces membrane pore formation, leading to membrane lysis. 37 Similar to GSDMD, GSDME can form transmembrane pores after cleavage. 38 HCC patients exhibit higher expression levels of GSDMD compared to normal tissue because the downregulation of GSDMD contributes to the attenuation of tumor proliferation via the intrinsic mitochondrial apoptotic pathway. 39 GSDMD is also demonstrated to suppress tumor growth by activating anti-tumor immunity, and therefore the lower expression of GSDMD is associated to poorer prognosis. 40 Although GSDME is traditionally regarded as a tumor suppressor, its upregulated expression in tumor tissues and in high-risk HCC patients suggests a potential tumor-promoting role, though the underlying mechanisms remain to be fully elucidated. Additionally, increased GSDME expression is associated with worse prognosis. 41 The proinflammatory cytokine interleukin-6 (IL-6) is upregulated by inflammasome-forming pattern recognition receptors (PRRs). 42 IL-6 modulates tumor immune microenvironment and promotes cancer development, and therefore higher expressions of IL-6 contribute to poorer prognosis. 43 IL-6 has been shown to activate T-cells, which in turn can enhance the process of epithelial-mesenchymal transition (EMT). In the context of HCC, previous research has identified biomarkers such as NOP16, which is implicated in mediating EMT through T-cell activation. 44 Nucleotide-binding oligomerization domain 2 (NOD2) is a member of the NOD-like receptor (NLR) family and induces pyroptosis. 45 NOD2 is demonstrated to promote carcinogenesis via pro-inflammatory response and programmed cell death, thus higher expressions of NOD2 is related to poorer prognosis. 46 The role of TIR domain-containing adaptor protein (TIRAP), another inflammatory mediator, in cancer remains unclear. Our findings suggest that key gasdermin family genes involved in pyroptosis may have dual roles in HCC tumorigenesis and progression.
Pyroptosis can restrict tumor development as a form of programmed cell death, whereas it also promotes tumor proliferation by assisting in the formation of a suitable microenvironment in certain biological contexts. 13 As indicated by our study, most of the pyroptosis-related genes are upregulated in HCC tissues compared to normal samples, and higher expressions of the selected genes were correlated with a poorer prognosis, indicating that pyroptosis might exert pro-tumor effects in HCC. In addition, crosstalks among forms of programmed cell death exist, and part of the pyroptosis-related genes might also involve in the regulation of other forms of programmed cell death, contributing to tumorigenesis and progression.
Previous study had reported that the GALAD (gender-age-Lens culinaris agglutinin-reactive alpha-fetoprotein-alpha-fetoprotein11 des-gamma-carboxy prothrombin) logistic regression model was established to diagnose hepatocellular carcinoma. 47 However, this model did not fully utilize more objective indicators, including genetic markers. Based on LASSO Cox regression, our study identified a pyroptosis-related signature and nomogram for predicting the survival of HCC patients. Compared to the conventional TNM stage, our nomogram demonstrates higher accuracy and sensitivity for prediction. The pyroptosis-based gene signature and nomogram provide evidence for the involvement of pyroptosis in HCC, and valid candidates for prognosis prediction and decision-making in clinical practice. 48
The functional enrichment analysis demonstrated that immune response pathways, complement pathways and cytokine activation pathways could be regulated by the pyroptosis-related genes. KIN001-220, TPCA-1, LY-303511, physostigmine, and vemurafenib are the top five potentially therapeutic drug candidates that could reverse the altered gene expressions in the high-risk group. Among the five drugs, LY-303511 can inhibit casein kinase, mammalian target of Rapamycin (MTOR), and phosphoinositide 3-kinase (PI3 K). As recent studies indicate that mTOR and PI3 K pathway is involved in pyroptosis, the LY-303511 could be a promising candidate to target the pyroptosis-related pathways in the high-risk group. 49 The new use for old drugs, for example, physostigmine, is also attractive and requires further investigation.
The immune context of tumor microenvironment is complex and diverse, and the discrepancy for the immune-cell infiltrations in different cohorts could be contributed by the dual effects of immune cells. Multifaceted roles of T cells have been identified in different types of cancer.50,51 Neutrophils in cancer exert different functions, which can promote cancer proliferation or tumor progression. 52
Previous studies have observed extensive immune cell infiltration in HCC tissue, predominantly consisting of T cells. These T cells have been proven to be associated with improved overall survival in HCC patients and with a lower rate of tumor recurrence following resection.53,54 Immunotherapy research has made significant progress in HCC, with developments such as PD-L1 inhibitors. 55 Since the infiltrated immune cells exert their effects in a context-dependent manner, their function in cancer is multifaceted and further studies are warranted to identify the role of infiltrated immune cells based on certain contexts.
Our study has several limitations. The datasets used did not include information on therapeutic responses, particularly to immunotherapy. Moreover, validation of the gene signature should be carried out through in vitro experiments and well-designed clinical studies. The predictive value of the signature also needs to be assessed using larger datasets from diverse populations. Despite these limitations, our research offers a comprehensive evaluation of pyroptosis-related genes in HCC, contributing valuable insights for future studies.
In summary, our research comprehensively evaluated pyroptosis-related gene profiles in hepatocellular carcinoma (HCC), leading to the development of a pyroptosis-gene signature comprising DHX9, GSDMD, GSDME, IL6, NOD2, and TIRAP. Additionally, we constructed a novel predictive nomogram that integrates this pyroptosis-gene-based risk score with clinicopathological features, enhancing prognostic accuracy. Functional enrichment analysis revealed significant correlations between pyroptosis-related genes and immune-related pathways. Moreover, these selected pyroptosis-related genes may influence immune cell infiltration and the composition of the tumor microenvironment, further underscoring their potential roles in HCC progression and immune modulation.
Conclusion
In conclusion, we performed a comprehensive analysis of pyroptosis-related genes in HCC, and constructed a pyroptosis-related gene signature with a predictive nomogram. Our results indicate that pyroptosis is important in HCC development and progression, and their essential function might be contributed by tumor immune microenvironment modulation. Potentially therapeutic drugs (KIN001-220, TPCA-1, LY-303511, physostigmine, vemurafenib, etc.) might alter the dysregulated pathways based on our in silico findings.
Supplemental Material
sj-pdf-1-thc-10.1177_09287329241296358 - Supplemental material for Predicting prognosis, immune landscape, and drug targets with a novel signature for hepatocellular carcinoma
Supplemental material, sj-pdf-1-thc-10.1177_09287329241296358 for Predicting prognosis, immune landscape, and drug targets with a novel signature for hepatocellular carcinoma by Xinqi Qiu, Wentao Huang, Jun Liang, Hao Chen, Weihong Sha, Yanlin Lyu, Kequan Chen, Hongwei Yang and Qingfang Zhang in Technology and Health Care
Footnotes
Author contributions
Conceptualization and design: XQQ, WTH, and QFZ, Collecting and assembly the data: XQQ, WTH, Data analysis and interpretation: XQQ, YLL, WHS, Visualization and validation: YLL, KQC and HC; Manuscript writing: XQQ, WTH, HWY, WHS and QFZ; All authors reviewed and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Medical Scientific Research Foundation of Guangdong Province of China (A2022412).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Code availability
R code used in this study are available from the corresponding author on reasonable request.
HZ revised the manuscript; all authors approved the final version of the article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.