
Abstract
Curing hepatitis C virus (HCV) infection in patients harbouring multiple severe comorbidities is a medical challenge. Evidence-based data are lacking regarding HCV treatment with direct-acting antiviral regimens in particular populations of HCV/HIV-coinfected patients with cirrhosis and chronic kidney disease on haemodialysis. Here, we present the HCV treatment challenges facing a patient with HIV coinfection, prior failure of both HIV-1 and HCV therapy, cirrhosis, end-stage renal failure on haemodialysis, as well as management of drug–drug interactions, especially given the need to receive long-term amiodarone therapy.
Introduction
Curing hepatitis C virus (HCV) infection in patients harbouring multiple severe comorbidities is a medical challenge involving expert teams from multiple medical specialties.
Here, we present a patient with HCV/human immunodeficiency virus (HIV) coinfection, cirrhosis, end-stage chronic kidney disease (CKD) on haemodialysis and heart disease on amiodarone therapy (clinical characteristics overview shown in Figure 1).
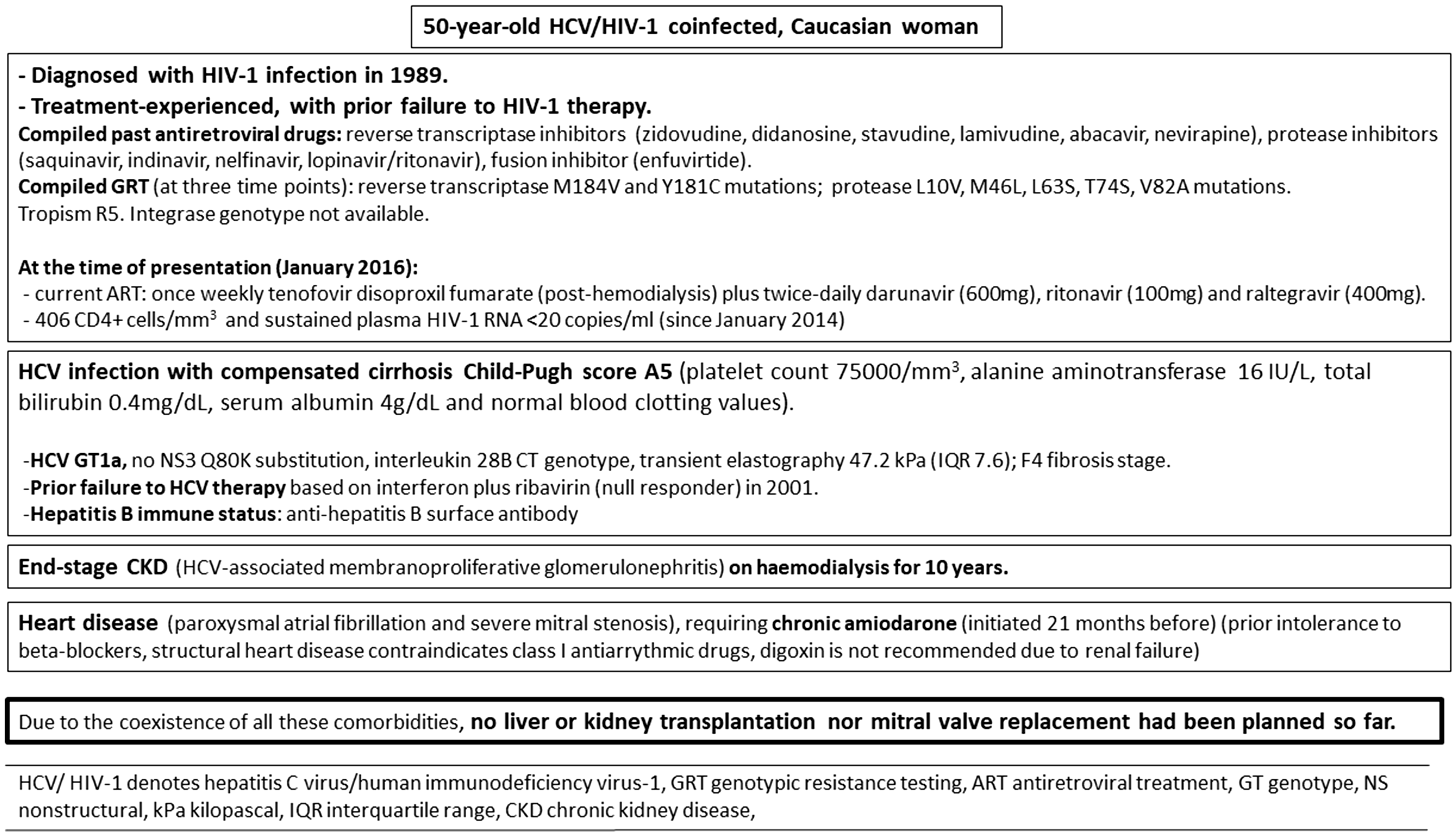
Baseline characteristics and comorbidities of the patient.
A direct-acting antiviral (DAA) regimen for the treatment of HCV was considered.
Pre-treatment considerations
Some issues were raised relating to HCV/HIV coinfection, with cirrhosis and severe CKD in the background, of the limited number of DAAs that have been trialled in this population.
A concerning drug–drug interaction (DDI) existed between DAAs and antiretroviral treatment (ART).
At the time of presentation, the patient received once-weekly tenofovir disoproxil fumarate (TDF) (post-haemodialysis) plus twice-daily darunavir (600 mg), ritonavir (100 mg) and raltegravir (400 mg). Her compiled drug-resistance genotype testing revealed reverse transcriptase M184V and Y181C mutations, conferring high-level resistance to lamivudine/emtricitabine and low-level resistance to abacavir, with all non-nucleoside analogues compromised. Protease sequencing revealed L10V, M46L, L63S, T74S, V82A mutations, without darunavir-associated mutations.
A study evaluated ombitasvir plus ritonavir boosted-paritaprevir plus dasabuvir (known as the 3D regimen) in HCV genotype 1 subjects with compensated cirrhosis and severe CKD (69% on dialysis). 1
Since no resistance-associated mutations to darunavir were present, the dose of darunavir could be reduced to 800 mg once daily, sharing the booster effect of ritonavir (100 mg once daily) over both paritaprevir and darunavir.
A pharmacokinetic study of HCV/HIV-coinfected patients on 3D revealed a decrease in darunavir minimum plasma concentration, without clinical significance in the absence of extensive protease inhibitor resistance. 2
On the contrary, the modification in plasma concentrations of 3D components, induced by darunavir in healthy volunteers, was not expected to affect 3D efficacy. Nevertheless, in a cohort of 22 HCV/HIV-coinfected patients who received 3D, the only two patients who failed to obtain a sustained virological response 12 weeks after ending treatment (SVR12) were on darunavir-based ART. 3
The United States Food and Drug Administration, 4 in contrast to the European Medicines Agency, 5 does not recommend the use of darunavir with the 3D regimen.
Another concerning DDI existed between 3D and amiodarone.
Post-marketing adverse event reports suggest a probable causal association between sofosbuvir and other DAAs (simeprevir, ledipasvir and daclatasvir) taken together with amiodarone, causing life-threatening bradycardia. 6
Data are lacking about potential bradycardia appearing when amiodarone is administered with 3D.
Nevertheless, it is worth noting that paritaprevir (nonstructural [NS]3/4A protease inhibitor), ombitasvir (NS5A inhibitor) and dasabuvir (non-nucleoside NS5B polymerase inhibitor) act as both substrates and inhibitors of P-glycoprotein (P-gp) and share similar mechanisms of action to simeprevir (NS3/4A protease inhibitor), ledipasvir and daclatasvir (both NS5A inhibitors) and sofosbuvir (nucleotide NS5B polymerase inhibitor), respectively.
Amiodarone is an inhibitor of P-gp and sofosbuvir is a substrate of P-gp. Amiodarone could increase the plasma concentrations of sofosbuvir. Of note, bradycardia has not been a recognized toxicity of sofosbuvir in clinical trials.
Furthermore, simeprevir, ledipasvir and daclatasvir have high protein binding like amiodarone. DAAs could displace amiodarone from its binding site and increase free (active) amiodarone concentrations and toxicity. 7
In addition, ritonavir is an inhibitor of P-gp and cytochrome (CYP) 3A4, leading to increased amiodarone exposure.
However, our patient had already been on ritonavir (100 mg twice daily)-boosted darunavir plus amiodarone for a long time without overt toxicity (no problems with heart rhythm, lung, thyroid or eyes).
Finally, ribavirin should be added (HCV genotype 1a and cirrhosis), resulting in greater potential for haematological toxicity due to reduced renal clearance.
Elbasvir and grazoprevir 8 and, recently, glecaprevir and pibrentasvir, 9 have demonstrated safety and efficacy in HCV-infected patients with cirrhosis and severe CKD. At the time when we considered to treat our patient, these DAAs were not available within Spanish public healthcare system.
Besides, coadministration of ritonavir-boosted darunavir and either of these newer DAAs is contraindicated. According to the compiled HIV drug-resistance genotype testing, darunavir should be necessarily included in ART for maintaining a suppressed HIV viral load. Moreover, plasma concentrations of amiodarone could increase as grazoprevir and glecaprevir are inhibitors of CYP3A4; therefore, toxicity monitoring of amiodarone would be required as well.
Furthermore, data are lacking in HCV/HIV-coinfected patients with cirrhosis and CKD on haemodialysis, and females are underrepresented in studies with all these regimens.
Treatment
Despite concerns, 3D plus ribavirin was deemed as the most reasonable treatment for the HCV infection.
Darunavir was reduced to 800 mg once daily, sharing the ritonavir (100 mg once daily) as a pharmacoenhancer included in the 3D regimen, TDF was maintained, and the integrase inhibitor raltegravir was switched to dolutegravir 50 mg once daily to reduce the ART regimen complexity.
In order to mitigate risks, close cardiac monitoring during the treatment was performed by electrocardiograms at baseline, over 24 h, 48 h, 7 days (steady state), 14 days, 21 days and 6 weeks. Neither bradycardia nor dizziness or syncope appeared during the complete course of the treatment (24 weeks). The overall tolerance was excellent without RBV-induced anaemia. HCV SVR12 was achieved and HIV-1 RNA remained <20 copies/ml during and after completion of the treatment.
Post-treatment considerations
This case highlights an HCV/HIV-coinfected patient with multiple and severe comorbidities, including cirrhosis, end-stage CKD on haemodialysis and heart disease. The treatment for HCV infection was challenging and its cure will pave the way to tackle subsequent treatment for the patient’s remaining diseases (including renal transplantation).
Close clinical monitoring of safety concerns and DDIs are imperative to manage this hard-to-treat population.
Informed consent
The patient provided written consent for the publication of her clinical record.
Footnotes
Authors’ contribution
HA and JML contributed on the concept and drafting of the manuscript. AM, NV, SK, SB, and JS contributed on the critical revision of the manuscript. All the authors approved the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Hortensia Álvarez has received support for attending meetings from Janssen-Cilag, Gilead Sciences, ViiV Healthcare, AbbVie, and Merck Sharp & Dohme. Ana Mariño has received support for attending meetings from Janssen-Cilag, Gilead Sciences, ViiV Healthcare, AbbVie and Bristol-Myers Squibb. Saye Khoo has received funding for the Liverpool Hepatitis Drug Interactions Website (![]() ) from Merck Sharp & Dohme, Janssen, Gilead Sciences and AbbVie, although editorial content remains independent. Sanjay Bhagani has received research support, speaker fees and travel grants from AbbVie, Bristol-Myers Squibb, Gilead Sciences, Merck Sharp & Dohme and ViiV Healthcare. Jonathan Schapiro has received research support, honorarium or consulting fees from the following: AbbVie, Merck Sharp & Dohme, Gilead Sciences, GlaxoSmithKline, Tibotec-Janssen, Bristol-Myers Squibb, Teva, Virology Education and ViiV Healthcare. He has received travel support and stipends for advisory work for the World Health Organization. Josep M Llibre has received support for attending meetings from Gilead Sciences, ViiV Healthcare, Janssen-Cilag, Bristol-Myers Squibb and Merck Sharp & Dohme. The remaining author declares no relevant conflicts of interest to the content of the manuscript.
) from Merck Sharp & Dohme, Janssen, Gilead Sciences and AbbVie, although editorial content remains independent. Sanjay Bhagani has received research support, speaker fees and travel grants from AbbVie, Bristol-Myers Squibb, Gilead Sciences, Merck Sharp & Dohme and ViiV Healthcare. Jonathan Schapiro has received research support, honorarium or consulting fees from the following: AbbVie, Merck Sharp & Dohme, Gilead Sciences, GlaxoSmithKline, Tibotec-Janssen, Bristol-Myers Squibb, Teva, Virology Education and ViiV Healthcare. He has received travel support and stipends for advisory work for the World Health Organization. Josep M Llibre has received support for attending meetings from Gilead Sciences, ViiV Healthcare, Janssen-Cilag, Bristol-Myers Squibb and Merck Sharp & Dohme. The remaining author declares no relevant conflicts of interest to the content of the manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.