
Abstract
Background
The global emergence of antimicrobial-resistant Neisseria gonorrhoeae represents a major public health concern. In particular, strains carrying the mosaic penA-60.001 allele have been associated with resistance to extended-spectrum cephalosporins and high-level resistance to azithromycin, threatening current treatment strategies. We describe an extensively drug-resistant Neisseria gonorrhoeae (XDR-NG) strain belonging to sequence type (ST) 16406, detected in Northern Italy in April 2025.
Methods
The N. gonorrhoeae strain was recovered from a urethral swab collected from a 28-year-old male at the Microbiology Unit of IRCCS Azienda Ospedaliero-Universitaria di Bologna. Antimicrobial susceptibility testing was performed using E-test strips and interpreted according to EUCAST breakpoints. Whole-genome sequencing (WGS) was conducted using Illumina technology, and genomic characterization included MLST, NG-MAST, NG-STAR typing, resistance gene detection, and phylogenetic analysis with publicly available ST16406 genomes.
Results
The isolate exhibited high-level resistance to tetracycline (MIC = 16 mg/L) and ciprofloxacin (MIC = 4 mg/L), very high azithromycin MIC (>256 mg/L), and resistance to both injectable and oral ESCs (ceftriaxone MIC = 0.25 mg/L, cefixime MIC = 1 mg/L). WGS assigned the isolate to MLST ST16406 and identified the mosaic penA-60.001 allele, the A2059G mutation in all four 23S rRNA alleles, mutations in gyrA and parC associated with fluoroquinolone resistance, and tetracycline resistance determinants including tet(M). The A39T mutation was identified in the mtrR repressor gene. No plasmids were detected using PlasmidFinder. Phylogenetic analysis demonstrated close genetic relatedness to previously reported ST16406 isolates from Europe and Asia.
Conclusions
This study reports the first detection in Italy of an XDR N. gonorrhoeae ST16406 isolate carrying the penA-60.001 allele. The finding highlights the ongoing international dissemination of this resistant lineage and underscores the importance of maintaining culture-based diagnostics to ensure effective antimicrobial resistance surveillance.
Keywords
Introduction
Neisseria gonorrhoeae (NG) is the etiological agent of gonorrhoea, one of the most common bacterial sexually transmitted infections (STIs) worldwide, with significant clinical and public health impact. 1 Genital infections typically manifest as urethritis in men and urethritis or cervicitis in women, although a wide spectrum of complications may occur, including epididymitis and prostatitis in men, endometritis and salpingitis in women, and disseminated gonococcal infection with systemic involvement. 1 Extragenital infections, particularly of the rectum and oropharynx, are frequent among men who have sex with men (MSM) reporting unprotected oral or anal intercourse. A substantial proportion of infections are asymptomatic, facilitating transmission, delaying diagnosis, and increasing the risk of complications. 2
NG has been classified by the World Health Organization (WHO) as a priority pathogen due to the rapid emergence of antimicrobial resistance (AMR), including multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains. 3 Owing to widespread resistance to fluoroquinolones and increasing resistance to azithromycin, third-generation cephalosporins, particularly ceftriaxone, represent the recommended first-line treatment. 3 However, resistance to extended-spectrum cephalosporins (ESCs) threatens current therapeutic strategies.
Molecular typing methods, including NG multiantigen sequence typing (NG-MAST), NG sequence typing for antimicrobial resistance (NG-STAR), and whole-genome sequencing (WGS), have become essential tools for surveillance and for predicting antimicrobial susceptibility patterns. 4 Genomic analysis provides valuable tools for the characterization of resistant lineages and supports surveillance efforts aimed at monitoring the spread of antimicrobial-resistant N. gonorrhoeae.
In recent years, several XDR NG isolates belonging to sequence type (ST) 16406 have been reported in Austria, the United Kingdom, France, Cambodia, and other countries.5–7 Here, we report the detection and genomic characterization of an XDR N. gonorrhoeae strain belonging to ST16406 identified in Italy in April 2025.
Materials and methods
Strain isolation and antimicrobial susceptibility testing
In April 2025, during routine diagnostics at the Microbiology Unit of IRCCS Azienda Ospedaliero-Universitaria of Bologna (Italy), an NG strain (designated CV23) was isolated from a urethral swab of a 28-year-old male.
The specimen (E-swab; Copan, Brescia, Italy) was inoculated onto modified Thayer–Martin agar (Kima, Piove di Sacco, Italy) and incubated at 37°C in 5% CO2 for 48 h. Presumptive NG colonies (oxidase-positive, greyish) were sub-cultured on chocolate agar under the same conditions. Species identification was performed by MALDI-TOF mass spectrometry (Bruker, Bremen, Germany).
Antimicrobial susceptibility testing (AST) was performed using E-test (bioMérieux, Marcy-l’Étoile, France) according to the manufacturer’s instructions. All MIC determinations were performed in duplicate to confirm the reproducibility of the results. Quality control procedures were performed using the recommended reference strain Neisseria gonorrhoeae ATCC 49226, and MIC values were within the expected quality control ranges.
Minimum inhibitory concentrations (MICs) were interpreted using EUCAST clinical breakpoints (version 16.0) for N. gonorrhoeae (available at https://www.eucast.org/clinical_breakpoints).
In addition to the recommended antimicrobials with established EUCAST clinical breakpoints or epidemiological cut-off values (ciprofloxacin, azithromycin, tetracycline, cefixime, ceftriaxone, and cefotaxime), additional agents (e.g., ertapenem, gentamicin, and doxycycline) were also tested to provide a broader characterization of the antimicrobial susceptibility profile of the isolate. Beta-lactamase production was assessed by nitrocefin test.
Genomic analysis
Whole-genome sequencing and genomic data analysis were carried out as follows. Genomic DNA was extracted by DNeasy Blood&Tissue Kit (Qiagen, Hombrechtikon, Switzerland), whereas Illumina paired-end libraries were generated using DNA Prep Library Preparation Kit (Illumina, San Diego, CA, USA). Sequencing was performed on an Illumina MiSeq platform (300 cycles, 2 × 150 bp).
Trimming of the short reads was performed with trim_galore v.0.6.10 with the automatic detection of the adapters and length of 50 (https://github.com/FelixKrueger/TrimGalore). Fastqc was used for the quality control of the raw and the trimmed reads (https://github.com/s-andrews/FastQC). De novo genome assembly and annotation were performed using SPAdes version 4.0.0 and Prokka version 1.14.6. QUAST version 5.3.0 was employed for the quality assessment of the assembled genome with the parameter --min-contig set to 0 and to 200. Sequencing depth and genome coverage were calculated by mapping the reads back to the assembly using BWA and the samtools coverage function from SAMtools.
In silico multi-locus sequence typing (MLST) was determined using the mlst command-line program against the PubMLST typing schemes (https://github.com/tseemann/mlst).
Further characterization of the sequence type was performed with NG-MAST (N. gonorrhoeae multiantigen sequence typing) and NG-STAR (N. gonorrhoeae sequence typing for antimicrobial resistance) directly from the PubMLST website (https://pubmlst.org/neisseria).
Antimicrobial resistance genes were identified using the NCBI Antimicrobial Resistance Gene Finder software AMRFinderPlus (https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/AMRFinder/). PlasmidFinder was used to detect the presence of plasmids (https://github.com/genomicepidemiology/plasmidfinder). The analysis was automated using a Snakemake script (v8.25.5).
To generate a phylogenetic tree, all isolates belonging to the same sequence type (ST16406) with publicly available genome sequences were retrieved from the PubMLST database. In addition, two recently reported French isolates (F95 and F96) were retrieved from NCBI and included in the analysis. Snippy v4.6.0 (https://github.com/tseemann/snippy) was then used for creating a core genome alignment against the FA1090 strain (ST1899), a laboratory reference strain. The generated alignment was then corrected for recombination by Gubbins v3.4 (https://github.com/nickjcroucher/gubbins/releases).
Pairwise single nucleotide polymorphism (SNP) distances were calculated from the recombination-filtered core genome alignment obtained after Gubbins correction using snp-dists (https://github.com/tseemann/snp-dists).
The resulting alignment was given as input for RAxML v8.2.12 (https://bioweb.pasteur.fr/packages/pack@RAxML@8.2.12). The phylogenetic tree was visualized using itol (https://itol.embl.de/).
Data availability
Raw sequencing data are available in GenBank under BioSample ID SAMN49969563 (BioProject PRJNA1291581). Reads were submitted to the Sequence Read Archive (SRA) (accession number SRR37461583).
Results
Phenotypic antimicrobial susceptibility
Antimicrobial susceptibility testing of N. gonorrhoeae CV23 strain. Minimum inhibitory concentrations (MICs) were interpreted using EUCAST clinical breakpoints (version 16.0) for N. gonorrhoeae.
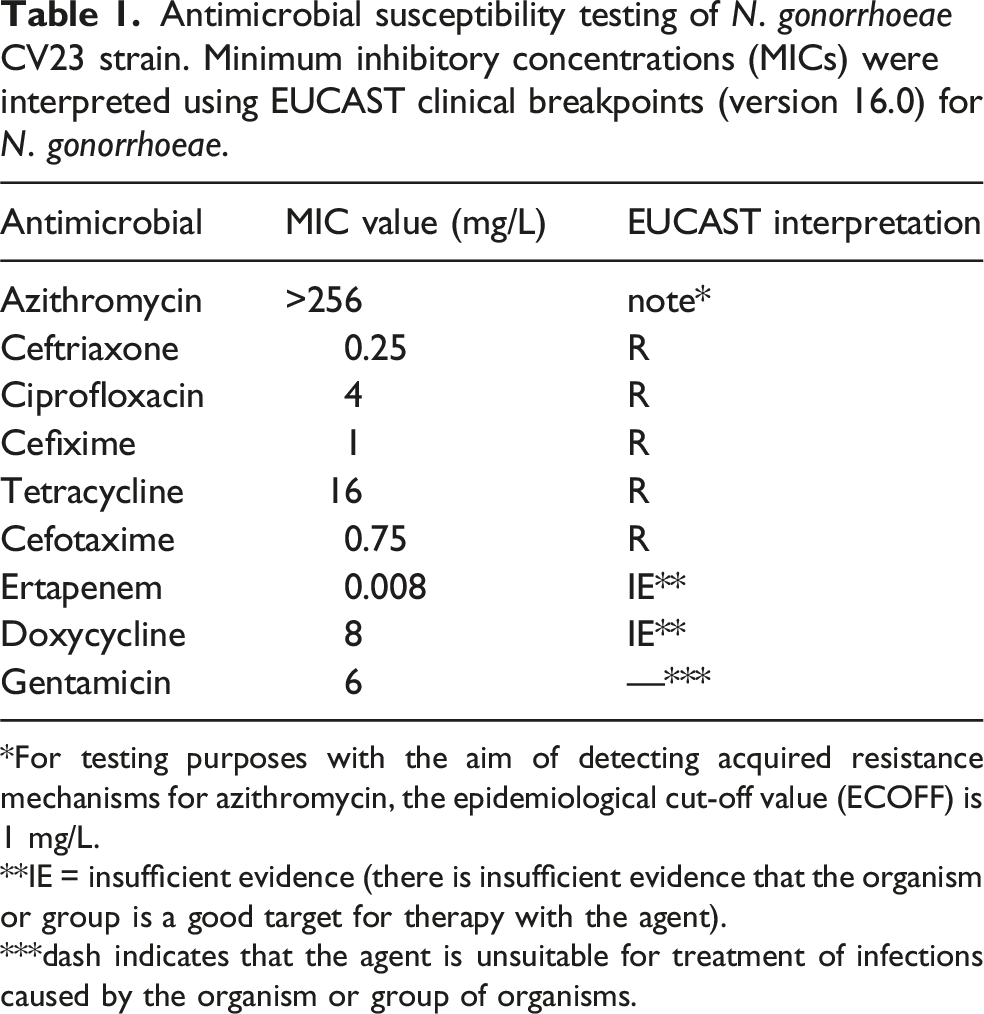
*For testing purposes with the aim of detecting acquired resistance mechanisms for azithromycin, the epidemiological cut-off value (ECOFF) is 1 mg/L.
**IE = insufficient evidence (there is insufficient evidence that the organism or group is a good target for therapy with the agent).
***dash indicates that the agent is unsuitable for treatment of infections caused by the organism or group of organisms.
According to the definition proposed by Tapsall et al. 8 the isolate was classified as XDR Neisseria gonorrhoeae based on resistance to injectable and oral ESCs and at least three additional antimicrobial classes. This classification has been widely used in surveillance studies and recent reports describing ceftriaxone-resistant ST16406 lineages. 9
The resistance phenotype was independently confirmed by the Italian National Institute of Health (Istituto Superiore di Sanità), the national reference laboratory for Neisseria gonorrhoeae antimicrobial resistance surveillance.
Genomic analysis
Illumina sequencing produced a total of 255 015 paired-end reads (2 x 150 bp). De novo assembly produced 426 contigs ranging from 78 to 172 497 bp in length, of which 160 contigs are longer than or equal to 200 bp. The total assembly length was 2 220 704 bp including all contigs and 2 188 583 bp when applying a minimum contig length threshold of 200 bp. The GC content was 52.3% and the N50 was 52 985 bp. Mapping of reads back to the assembly indicated an average sequencing depth of 33.6x with 99.7% breadth of coverage. Taxonomic classification showed that 95.99% of reads were classified. Abundance re-estimation assigned 99.04% of the classified reads to Neisseria gonorrhoeae, indicating high sample purity.
MLST assigned CV23 to ST16406. NG-MAST and NG-STAR typing identified ST22862 and profile 5793, respectively. No plasmids were detected using PlasmidFinder; however, the fragmented nature of short-read assemblies may limit the detection of small or incomplete plasmid sequences.
Antibiotic resistance genes and mutations identified in N. gonorrhoeae CV23 strain.
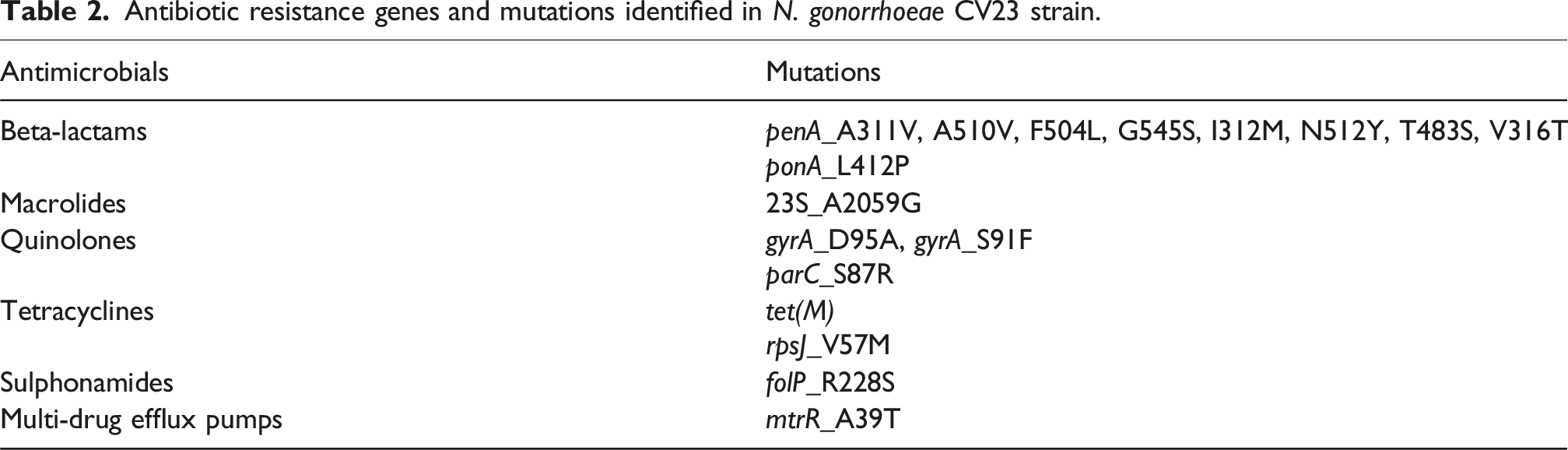
Quinolone resistance-determining regions (QRDRs) showed S91F and D95A substitutions in gyrA (a subcomponent of DNA gyrase) and S87R in parC (a component of topo-isomerase IV), consistent with ciprofloxacin resistance. Tetracycline resistance determinants included the tet(M) gene and the V57M substitution in rpsJ (encoding the S10 ribosomal protein). The A39T mutation was identified in the mtrR repressor gene.
We aligned the genome of CV23 with 15 extensively resistant strains belonging to the same ST previously isolated in Europe (France, UK, Austria, Norway), Cambodia and Australia.
Phylogenetic analysis demonstrated close relatedness to previously reported ST16406 isolates (Supplemental Figure 1). CV23 differed by 27 single nucleotide polymorphisms (SNPs) from Cambodian strain 22R655567T and by 32–35 SNPs from two French isolates (F95 and F96), indicating close genetic relatedness within the ST16406 lineage.
Discussion
We report the first detection in Italy of an XDR N. gonorrhoeae strain belonging to ST16406 and exhibiting resistance to ESCs. Previous Italian surveillance data had not documented ceftriaxone-resistant isolates, 10 highlighting the epidemiological relevance of this finding.
Genomic analysis revealed high similarity to ST16406 strains recently reported in France and Cambodia. 6 The limited SNP distance (approximately 30 SNPs) is consistent with the international dissemination of this resistant lineage. However, while indicating close genetic relatedness, the available genomic data do not support inference of direct transmission events.
The presence of the mosaic penA-60.001 allele represents a key determinant of resistance to ESCs. This allele encodes structural alterations in penicillin-binding protein 2 that decrease affinity for cephalosporins. Additional chromosomal mutations, including the A39T substitution in the mtrR repressor gene, have been associated with increased expression of the MtrCDE efflux pump and may contribute to elevated MICs in combination with other resistance determinants. 11
High azithromycin MIC was explained by the A2059G mutation in all four 23S rRNA alleles, a well-recognized mechanism associated with MICs exceeding 256 mg/L. Ciprofloxacin resistance was attributable to classical QRDR mutations in gyrA and parC. 12 Tetracycline resistance was primarily mediated by the tet(M) gene, while the rpsJ V57M substitution may contribute to the resistance phenotype.
Most of these resistance determinants, including mosaic penA alleles and mutations in gyrA, parC, and mtrR, are believed to originate from horizontal gene transfer events involving commensal Neisseria species inhabiting the oropharynx. 3 This highlights the role of pharyngeal reservoirs in the evolution of antimicrobial resistance.
Unlike some previously described ST16406 isolates, 6 CV23 did not carry beta-lactamase genes such as blaTEM. This finding was corroborated by the absence of plasmids and by the phenotypic absence of production of beta-lactamases (nitrocefin test).
It should be acknowledged that the genome assembly obtained from short-read sequencing was fragmented, which is common for N. gonorrhoeae genomes generated using Illumina technology. Although this approach allows reliable identification of most resistance determinants, it may limit the detection of structural genomic elements, including small or fragmented plasmids.
Another major limitation of this study is the absence of detailed clinical, demographic, and behavioral data, including sexual orientation (e.g., MSM status), travel history, and contact tracing information. Moreover, the absence of data on treatment and test-of-cure also prevents assessment of the clinical significance of the observed ceftriaxone MIC in this case.
Therefore, the origin of the strain, its possible route of introduction into Italy, and its transmission network cannot be determined.
Several ceftriaxone-resistant NG cases detected in Europe have been associated with travel or sexual contacts in the Asia-Pacific region, where penA-60.001-associated lineages appear to be more prevalent.5–7 In the absence of travel or contact information for the present case, this epidemiological context may be relevant for interpreting the detection of this lineage in Italy.
Furthermore, despite being categorized as resistant according to EUCAST breakpoints, some infections caused by penA-60.001 strains have been successfully treated with higher-dose ceftriaxone regimens. Consequently, the clinical significance of this resistance phenotype may vary, and cannot be assessed in the present case due to the absence of treatment and follow-up information.
In conclusion, even though the origin of the CV23 strain cannot be determined, this report describes the microbiological detection and genomic characterization of an XDR N. gonorrhoeae ST16406 isolate identified in Italy.
The increasing reliance on nucleic acid amplification tests (NAATs), including home-based diagnostic approaches, may further reduce the availability of cultured isolates for antimicrobial susceptibility testing. Therefore, maintaining culture capacity alongside molecular diagnostics remains essential for effective surveillance of antimicrobial resistance in N. gonorrhoeae at both European and global levels.
Continuous microbiological surveillance combined with genomic epidemiology is essential to monitor dissemination of resistant lineages, inform treatment guidelines, and prevent further spread of untreatable gonococcal infections.
Supplemental material
Supplemental Material - Detection and genomic characterization of an extensively drug-resistant Neisseria gonorrhoeae ST16406 isolate identified in Italy, 2025
Supplemental Material for Detection and genomic characterization of an extensively drug-resistant Neisseria gonorrhoeae ST16406 isolate identified in Italy, 2025 by Raul Cetatean, Benedetti Secci, Tiziana Lazzarotto, Claudio Foschi, Simone Ambretti in International Journal of STD & AIDS
Footnotes
Acknowledgments
We would like to thank the Clinical Laboratory Scientists of the Bacteriology section of the Microbiology Unit of IRCCS Policlinico Sant'Orsola in Bologna for the assistance during the study.
Ethical considerations
This study was carried out in accordance with the declaration of Helsinki, under the terms of relevant local legislation, and was cleared by the institutional review Board. The requirement for informed consent was waived due to the observational nature of this study.
Author’s contribution
SA, CF, and TL conceived and designed the study. RC and BS performed the experiments and analysed the data. CF and RC wrote the paper. All authors read, reviewed and approved the final version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work received no specific grant from any funding agency, and it was supported by internal funding. RC was supported by EU funding within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project no. PE00000007, INF-ACT).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.