
Abstract
Background
Hereditary vestibular dysfunctions (HVDs) are a group of diseases caused by genetic mutations, characterized by congenital or progressive vestibular dysfunction, often accompanied by hearing loss or other systemic damages. These diseases are divided into syndromic (e.g., Usher syndrome, CHARGE syndrome) and non-syndromic types, involving mutations in key genes such as MYO7A, COCH, SLC26A4, TMC1, etc. Although clinical phenotypes vary, the pathogenesis is complex, traditional diagnostic methods are limited, and effective treatments are lacking. Mouse models are important tools for studying hereditary vestibular dysfunction, providing critical platforms for understanding disease mechanisms, developing diagnostic biomarkers, and treatment strategies.
Methods
This review systematically searched English and Chinese literature in databases including PubMed, Web of Science, Embase, and CNKI from January 2000 to April 2026. The search strategy combined Medical Subject Headings (MeSH) terms and free-text keywords, including “hereditary vestibular dysfunction,” “mouse models,” “gene therapy,” “CRISPR-Cas9,” “Usher syndrome,” “translational research,” “biomarkers,” “Meniere disease,” and “International Mouse Phenotyping Consortium.” Inclusion criteria were: (1) peer-reviewed articles on hereditary vestibular dysfunction mouse models; (2) studies reporting genetic mechanisms, pathophysiology, or therapeutic interventions; and (3) English or Chinese language publications. Exclusion criteria were: (1) non-peer-reviewed conference abstracts or preprints and (2) studies without clear genetic or phenotypic characterization. Two authors independently screened titles, abstracts, and full texts, with disagreements resolved by consensus. The review focuses on analyzing the applications of spontaneous mutation models, genetic engineering models, CRISPR technology-based models, and knockout models from the International Mouse Phenotype Consortium (IMPC) in disease mechanism research and treatment development.
Results
In recent years, significant progress has been made in hereditary vestibular dysfunction mouse model research. Spontaneous mutation models like Myo6 and Cdh23 mutant mice have revealed the key role of cytoskeletal and cell junctions in vestibular function. Genetic engineering models have successfully simulated a variety of diseases, including Usher syndrome, ion channel defects, and vestibular development abnormalities, elucidating the molecular mechanisms of TMC1/2 mechanosensory channels, SLC26A4 ion transport, and vestibular system development genes. The application of CRISPR-Cas9 technology has greatly improved model construction efficiency and precision. These models have shown positive results in gene therapy, gene editing, and drug treatment research. AAV-mediated gene replacement therapy, CRISPR gene repair, and new drugs such as α1-antitrypsin have all achieved positive outcomes in mouse models. Biomarker studies based on multi-omics techniques have identified potential diagnostic markers such as Slc17a6 and BDNF.
Conclusion
Mouse models play an irreplaceable role in the study of hereditary vestibular dysfunction, providing a solid foundation for elucidating disease mechanisms, improving diagnostic methods, and developing treatment strategies. Although clinical translation still faces challenges such as species differences, delivery efficiency, and treatment windows, with the continuous development of gene editing technology, nano-delivery systems, and multi-omics techniques, personalized diagnosis and treatment for hereditary vestibular dysfunction are expected to be realized, bringing new hope to patients.
Keywords
Introduction
The vestibular system is a key sensory organ located in the inner ear, crucial for maintaining balance, spatial orientation, and coordinating head and eye movements. It includes the semicircular canals and otolith organs (utricle and saccule), which detect angular and linear accelerations. By integrating with visual and proprioceptive signals, it ensures postural stability and visual fixation during movement. Vestibular dysfunction leads to severe symptoms such as vertigo, balance disorders, and nystagmus, significantly affecting quality of life. 1
Hereditary vestibular dysfunctions (HVDs) are diseases caused by gene mutations, characterized by congenital or progressive vestibular impairment, often accompanied by hearing loss or other systemic abnormalities. These disorders are divided into syndromic types (e.g., Usher syndrome, Pendred syndrome, and DFNA9, which involve additional sensory or organ abnormalities) and non-syndromic types (primarily manifesting as balance issues). Mutations in genes such as MYO7A, COCH, SLC26A4, and OTOF disrupt hair cell function, mechanotransduction, or ionic homeostasis.2,3 Epidemiologically, HVDs are rare, with Usher syndrome affecting about 1 in 10,000 people, and some autosomal recessive forms as low as 1 in 500,000. Due to phenotypic heterogeneity and diagnostic limitations, they are often underdiagnosed.4,5
Mouse models are indispensable for studying HVDs. By simulating human gene mutations—such as natural mutations in Myo7a and Cdh23, and genetically engineered or CRISPR-Cas9 models for Slc26a4 and Coch—researchers have uncovered mechanisms like defective mechanotransduction, ionic imbalance, and cytoskeletal abnormalities.6,7 These models are evaluated through behavioral tests (e.g., rotarod tests) and electrophysiological assessments (e.g., vestibular evoked myogenic potentials), supporting preclinical trials in gene and drug therapy.
Although differences between mouse and human vestibular anatomy and physiology limit direct clinical translation, these models have laid the groundwork for understanding disease mechanisms and developing precision medicine. Previous reviews have examined the genetic bases of vestibular disorders, including systematic analyses of genes linked to hearing and vestibular phenotypes in humans and mice8,9 and mini-reviews on molecular and genetic mechanisms of vestibular disorders. 10 Building upon these works, the present review specifically focuses on summarizing recent advances in HVDs mouse models, explores their roles in studying pathogenesis, biomarker identification, and treatment development, analyzes the challenges of clinical translation, and envisions personalized treatment prospects through integrated genomics and human studies.
Vestibular system: Anatomy and physiological basis
Anatomical structure of the vestibular system
The vestibular system is the core structure in the inner ear responsible for balance and spatial orientation. It resides within the bony labyrinth of the petrous part of the temporal bone. The bony labyrinth includes the vestibule and three semicircular canals (anterior, posterior, and lateral), while the membranous labyrinth is suspended inside, supported by delicate connective tissues, and contains the motion-sensing components: the utricle, saccule, and the three semicircular ducts. 1 The semicircular canals are arranged approximately orthogonally and detect rotational head movements (angular acceleration). Each canal has an enlarged area called the ampulla, which contains a sensory structure known as the crista ampullaris. The stereocilia of hair cells in this crest are embedded in a gelatinous structure called the cupula. When the head rotates, endolymph within the canals lags behind due to inertia, deflecting the cupula and stimulating the hair cells to generate neural signals.
The utricle and saccule—collectively called the otolith organs—sense linear acceleration and head position relative to gravity. These organs contain maculae with hair cells whose stereocilia are embedded in a gelatinous layer topped with otoconia (calcium carbonate crystals). When the head tilts or accelerates linearly, the otoconia shift, bending the stereocilia and initiating signal transduction. The vestibular nerve, a branch of the vestibulocochlear nerve (cranial nerve VIII), transmits these signals to the vestibular nuclei in the brainstem and to the cerebellum, where they integrate with visual and proprioceptive information to maintain balance and posture. 11
The bony labyrinth is filled with perilymph, while the membranous labyrinth contains potassium-rich endolymph, essential for hair cell depolarization. Hereditary vestibular dysfunctions are often caused by structural or functional abnormalities in hair cells or disruptions to the endolymphatic system. For instance, MYO7A mutations affect stereocilia organization, while SLC26A4 mutations lead to ionic imbalance—underscoring the importance of anatomical research in understanding disease mechanisms and developing treatments. 12
Physiological functions of the vestibular system
The vestibular apparatus is very important for keeping body balance, making vision stable and knowing space position, which mainly depends on two basic reflexes named vestibulo-ocular reflex (VOR) and vestibulo-spinal reflex (VSR). 13 When head moves, VOR helps eyes move opposite direction to see things clearly without shaking, this happens because semicircular ducts sense head turning speed then let eyes adjust for steady view, it is especially needed when people walk or read. For example, when you quickly turn your head to one side, the VOR moves your eyes in the opposite direction to keep your vision stable. 14 The VSR maintains posture and balance by regulating muscle tone in response to head position and movement. The otolith organs detect linear acceleration and head tilt, sending signals to the spinal cord to adjust body posture. This reflex is critical for standing, walking, and running. For instance, when walking on uneven ground, the VSR helps stabilize your posture by adjusting leg and trunk muscle activity. 15
Beyond reflexes, the vestibular system provides continuous information about the position and movement of the head in space, which is integrated with visual and proprioceptive inputs in the brain to support spatial memory and navigation. Accurate spatial perception is essential for complex tasks such as driving or dancing.13,14 Dysfunction of the vestibular system can result in vertigo, imbalance, and nystagmus, severely impacting daily activities such as walking or reading. Research find the vestibular system can affect body’s automatic functions like heart and blood pressure control, also thinking ability such as memory for finding way, showing its many physiological roles. Knowing how it works is very important for developing accurate treatments, for example, drugs to treat DFNA9 sickness caused by COCH gene mutation. 15
Molecular mechanisms of vestibular system development
The growth of vestibular apparatus is a very complicated process, which needs many molecular signals to cooperate carefully for ensuring the hearing parts can develop in right way. The induction of otic placode marks the beginning of this process, where the transcription factors PAX2 and GATA3 are controlling to make otic vesicle form properly. According to Wu and Kelley in 2012, the auditory vesicle will later develop into semicircular ducts as well as utriculus and sacculus. The formation of auditory vesicle involves patterning and shape changes, in which FGF10 and Wnt signaling pathways are playing key roles. FGF10 helps the cristae and ducts of semicircular canal to grow better, while Wnt signaling is deciding how the sensory epithelium will develop. 16 The Notch pathway is responsible for differentiating hearing cells into different types, using lateral suppression to make progenitor cells become hair cells or support cells nearby. If this pathway has problems, it may cause too few or too many hair cells, for example, when Notch signaling not working properly can reduce vestibular hair cells number. 17 The SHH signaling pathway controls the ventral growth of otic vesicle, ensuring the inner ear can be correctly divided into different regions. Disruption of SHH signaling may lead to malformation of inner ear structures. The SOX2 transcription factor maintains progenitor cells survival and marks sensory areas, but ATOH1 is crucial for hair cells development, and its absence will cause severe defects in balance organs like semicircular canals or otolithic parts.16,18 The MYO7A gene is essential for stereocilia formation and its sensing function, but genetic mutations such as in “Usher syndrome” can disrupt stereocilia structure and cause balance disorders. For example, mice lacking Myo7a show disordered stereocilia arrangement and worsened vestibular function, as Bok J et al. 17 reported that mouse models with same gene changes provide useful understanding about growth disorders and is key for finding treatments, even using gene ways to repair hearing cell function. Correct handling of these tiny molecules matter a lot since it make vestibular system perform properly. Inherited balance problems come from genetic issues, so molecular research must be done to create accurate therapies.
Classification and genetic basis of hereditary vestibular dysfunction
Syndromic vestibular dysfunction
Syndromic vestibular disorders often make patients have balance issues along with other body problems, especially hearing and vision damages, and usually connect with many genetic diseases. Usher syndrome is the most typical case. This inherited sickness, requiring two faulty genes to happen, leads to born deafness, gradually worsening eye condition named retinitis pigmentosa, and sometimes balance troubles in patients. Usher syndrome is classified into three clinical types as follows. USH1 is associated with severe congenital deafness and complete vestibular areflexia, USH2 involves milder hearing loss with minimal or no vestibular symptoms, USH3 is characterized by progressive auditory and vestibular impairment. USH1 is caused by mutations in genes such as MYO7A, CDH23, PCDH15, USH1C, and USH1G, which encode proteins essential for the structural integrity and function of hair cells. Mutations lead to vestibular hair cell dysfunction. 19
In addition, CHARGE syndrome is a rare autosomal dominant disorder caused by mutations in the CHD7 gene. Patients exhibit multi-system developmental abnormalities, including auricular malformations, semicircular canal dysplasia, and vestibular dysfunction. CHD7 encodes a chromatin remodeling protein critical to inner ear development. Its deficiency results in a reduction of vestibular ganglion cells and structural malformations. 20 Genes implicated in syndromic disorders are often developmental regulators involved in inner ear morphogenesis and neural circuit formation, reflecting the complex genetic landscape underlying hereditary vestibular dysfunction.
Non-syndromic vestibular dysfunction
Non-syndromic vestibular dysfunction refers to hereditary conditions characterized solely by vestibular impairment without systemic or multisensory involvement. These disorders exhibit considerable genetic heterogeneity. Commonly implicated genes include GJB2, MYO7A, and SLC26A4. The GJB2 gene encodes connexin 26, one of the most prevalent causative genes of autosomal recessive sensorineural hearing loss. Although primarily associated with cochlear dysfunction, studies have shown that GJB2 is also expressed in vestibular organs. Some patients exhibit vestibular hypofunction—especially involving the saccule—while the semicircular canals and utricle often remain functionally intact.4–6 MYO7A mutations can cause both syndromic Usher syndrome and non-syndromic hereditary deafness (e.g., DFNB2). In non-syndromic forms, patients typically do not present with retinal degeneration or overt vestibular symptoms.7,11
Mutations in SLC26A4 are a major cause of non-syndromic vestibular dysfunction. These mutations commonly result in enlarged vestibular aqueduct (EVA) and are associated with both auditory and vestibular abnormalities. SLC26A4 encodes pendrin, a protein involved in inner ear ion homeostasis. Dysfunctional pendrin disrupts endolymphatic fluid metabolism, thereby impairing vestibular function. 13 Other genes, such as POU4F3 and USP48, have also been implicated in non-syndromic vestibular dysfunction, indicating that the genetic architecture of these conditions is both complex and diverse.14,15,18
Molecular pathological mechanisms of vestibular dysfunction
The molecular pathology causing inherited vestibular disorders have various defects, such as vestibular parts not developing right, hair cells failing to properly do mechanotransduction, nerve signals having issues, and also synapse transmission not working normal. Firstly, MYO7A and similar genes produce proteins that are very crucial for hair cell structure and its movement ability. Mutations in these genes lead to vestibular hair cells malfunctioning and structural defects, impacting how vestibular signals are generated and transmitted.
16
Moreover, changes in GJB2 gene encoding connexin 26 can disturb ion and signal exchange between inner ear cells, especially potassium flow, making hair cells’ electrical activity weaker and balance disorders occurs as Bok, it was said that the SLC26A4 gene changes can influence pendrin protein’s working, this protein has big job to control ion balance in inner ear fluid. When this protein does not do its job well, the vestibular canals will grow larger and bring balance trouble because the endolymph balance got disorder.
21
Also, the ribbon synapses inside vestibular hair cells need proteins like otoferlin to handle neurotransmitter release, if these proteins are not enough then vestibular nerve signals have difficulty passing through. But some animal experiments show there maybe exist compensation ways, this could explain why clinic symptoms appear different.
22
Besides, the neuroplasticity and adaptive reorganization in central nervous system also take part in vestibular disorders’ pathological mechanisms; such important changes can affect not just disease symptoms but also treatment effects. In short, the genetic causes of inner ear balance disorder come from many biological problems that influence each other, to deeply know these processes is very key for developing right treatment methods (Figure 1). Classification and molecular pathogenesis of HVDs.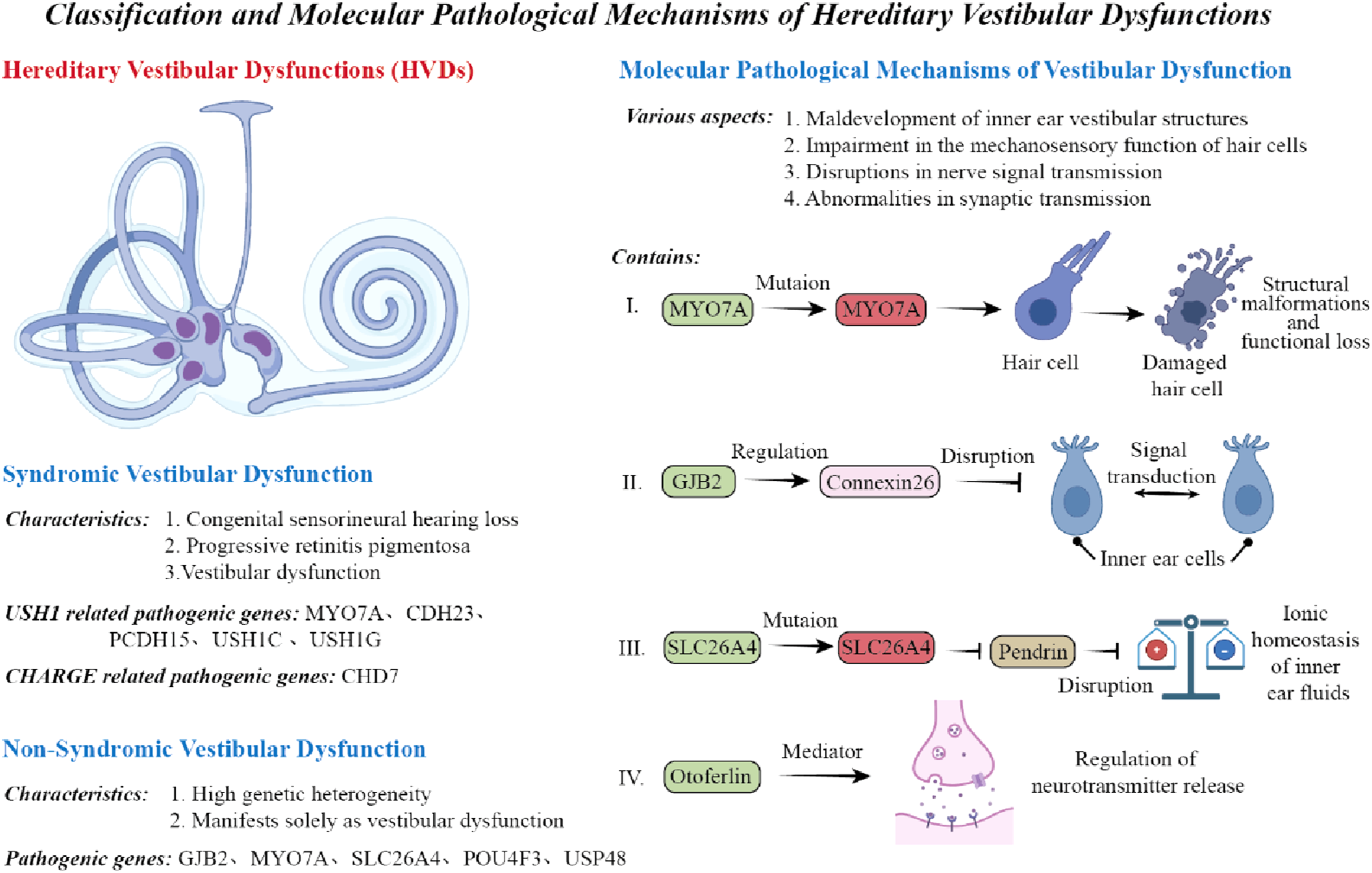
Methods for establishing mouse models of vestibular dysfunction
Spontaneous mutation mouse models
Spontaneous mutant mice strains naturally get genetic changes during breeding process, these changes can pass disease-like traits same as human conditions. Such models are very good for studying balance disorders since they show true genetic mutations and how these affect balance abilities. Usually, these mutations contain single DNA changes, gene deletions or insertions that harm protein structure or function, causing balance problems, for example, mice with Myo6 gene mutations show serious balance disorders and also hearing loss. Myo6 encodes an unconventional myosin involved in cytoskeletal organization and mechanosensory function of hair cells. Mutations in this gene lead to hair cell disorganization and impaired function of vestibular receptors, manifesting as head tilting, circling behavior, and balance disturbances. 23
Similarly, Cdh23 mutant mice—another spontaneous mutation model—have been extensively studied. CDH23 encodes a calcium-dependent adhesion molecule critical for hair cell mechanotransduction and intercellular junctions. If this gene sequence has changes, the auditory hair cells cannot work well, so hearing and balance will both get problems. For studying these problems, scientists using behavior tests, tissue checks, and electrical measurements to find out how inherited balance disorders happen and which molecular pathways involved. Spontaneous mutation models is good since they got natural gene patterns and can copy complex genetic diseases, some mutations are much like human disease mutations, this help to discover new disease genes and pathways. But also have bad sides such as mutations appear randomly, need much time to find out, and sometimes lead to death in development or just show small problems, so these models not widely used.23,24 Nowadays, modern gene sequencing tech makes finding and studying spontaneous mutations more easy, which helps better to create and check these models. Moreover, the mouse models having natural navigation mutations was used for studying vestibular disorders, where researchers carried out rotation tests, behavior observations, fluorescent labeling and gene analysis, finding that Slc17a6 may play key role in spatial processing, which give new insights about molecular pathways. 25
Genetically engineered mouse models
Genetically modified mouse models are created by molecular methods to delete, add or alter specific genes, making animals with precise gene changes that can mimic human inner ear diseases. These systems can accurately control gene activity and are very useful for investigating gene functions and disease mechanisms in the vestibular system, serving as key tools for studying genetic vestibular disorders. 26 For example, the deletion models targeting Usher syndrome-related gene USH1G exhibit severe hearing and balance issues, similar to human USH1G variants, and gene therapy in these animals has shown potential to restore balance functions, proving the clinical value of GM models.27,28 Furthermore, conditional gene targeting techniques like Cre-LoxP allow spatial and temporal control of gene deletion, preventing embryo death from whole-body deletions and enabling study of vestibular-specific gene roles. 29 Knock-in methods introduce human disease mutations to observe their exact effects on the balance system, while GM mouse models offer benefits like uniform genetic backgrounds and controlled mutation types, aiding mechanism research and treatment evaluation. However, creating these models requires significant time and cost, and genetic redundancy may mask observable traits, necessitating multiple models for comprehensive study due to varying mouse strain genetics. In recent years, GM models have become crucial for vestibular disorder research, advancing gene therapy and precision medicine.27–29 These frameworks are vital for basic research and provide the optimal platform to test the efficacy and safety of new genetic treatments, as seen in USH1G knockout mice where AAV gene therapy showed strong clinical potential. Additionally, targeted gene deletions help researchers examine gene roles across development stages and body regions, clarifying when and where vestibular diseases arise.
Application of CRISPR-Cas9 technology in the construction of vestibular dysfunction models
In recent years, CRISPR-Cas9 gene editing technology has achieved significant progress, because it can modify DNA sequences with high precision and efficiency. Compared to traditional genetic methods, CRISPR-Cas9 is more accurate, faster for creating samples, and cheaper, so now it is widely used to produce animal models with inherited inner ear diseases. In the CRISPR-Cas9 system, the guide RNA (gRNA) acts like a GPS to direct Cas9 enzyme to target specific DNA regions, then makes Cas9 cut both DNA strands. The damaged DNA is then repaired by either NHEJ or HDR pathways, allowing precise gene modifications such as disruption, insertion, or editing. For example, researchers using CRISPR-Cas9 have successfully altered the Tmc1 gene, which controls hair cell mechanotransduction, and created models mimicking human hearing loss. 30 These animals exhibit severe hearing and balance issues, making them ideal for testing gene therapies as reported by Nist-Lund et al. 31 Additionally, CRISPR enables development of mouse models carrying human disease mutations. These systems simplify direct study of mutation effects and treatment responses, advancing personalized medicine. 31 Beyond standard CRISPR-Cas9, newer tools like base editors and prime editors achieve even finer genome editing. These methods introduce single-base changes without DNA breaks, reducing off-target risks and improving clinical suitability. 32 Despite its advantages for inner ear research, CRISPR faces challenges like unintended gene edits, inefficient DNA repair, immune reactions, and delivery difficulties to ear tissues. Both viral vectors like AAV and synthetic carriers such as lipid nanoparticles are being refined for better precision and safety. Overall, CRISPR-Cas9 provides a robust platform for vestibular disorder modeling, offering new opportunities to investigate gene roles, disease mechanisms, and therapies. 23 It holds great potential for advancing customized treatments for genetic inner ear conditions.
IMPC knockout mouse models of vestibular dysfunction
Summary of representative mouse models of hereditary vestibular dysfunction.

Representative mouse models of hereditary vestibular dysfunction and research progress
Mouse models related to Usher syndrome
Usher syndrome (USH) is a genetic disorder mainly leading to hearing loss, retinitis pigmentosa, and balance issues, which is a major cause of deaf-blindness and has three types. USH1 shows serious deafness since birth and severe balance problems, while USH2 appears moderate hearing loss with nearly no balance trouble, and USH3 develops slowly in both hearing and balance aspects. The disease relates to genes such as USH1C, MYO7A, CDH23 and PCDH15, where mouse models help study its mechanism and find treatments, since USH1C produces harmonin protein crucial for ear hair cells’ structure and function. Mice having Ush1c gene mutations display disordered ear hairs and balance problems observed through poor movement control, 33 but injecting USH1C gene into their ears via AAV therapy significantly improved hearing and balance, indicating promising treatment effects. 27
The mice without MYO7A gene also have big hearing and balance trouble, same as those USH1B patients. The protein MYO7A plays key role in transporting stuff and feeling mechanical signals in stereocilia, when gene has mutation, hair cells will not function correct. 34 When CDH23 and PCDH15 genes got problem, the links between stereocilia will be broken, and then cause machinery not work good and balance control become bad. 24 Study on USH mouse models not only help know gene roles but also apply behavior tests, VsEP, and VOR to examine balance ability, which make better diagnosis and treatment ways. Various USH gene mutations lead to different disease pathways, this is helpful for making specific treatments. 35 Also, the CRISPR/Cas9 tool can very accurately modify genes linked to USH, which helps to quicker make various mouse models and push forward research on mechanisms and treatments. 28 Later on, by using single-cell sequencing plus multi-omics together, it can better show how USH genes are controlling the balance systems, which helps making personalized medicine development.
Ion channel–related mouse models of vestibular dysfunction
Ion channels is very important for keeping the electrochemical balance inside inner ear and also helping hair cells to do their sensing job, because vestibular apparatus can only work properly when many ion channels operate together, so if any channel have problems then vestibular disorders may happen. Recently, mouse models targeting ion channel genes has provided useful insights for understanding the molecular reasons of hereditary vestibular disorders, and TMC1 and TMC2, which is the proteins for mechanosensory channel, are thought to be the key parts in hair cell’s mechanotransduction system that cannot be missing. Changes in Tmc1 gene often cause hereditary deafness for human people, and mice lacking Tmc1 shows much lower VsEP and worse VOR, especially affecting how semicircular canal and balance organs works, while compared with Tmc2 knockout mice, those lacking Tmc1 shows more serious balance problems, which means Tmc1 plays bigger role in vestibular function. Moreover, studies show that using Tmc1 gene replacement therapy can help restore some balance functions; this gives a theoretical support for future genetic treatments. 36 The P2X2 receptor is a ion channel activated by ATP, it has important function for cells to talk with each other inside the inner ear, and mice having P2rx2 mutations shows problems in vestibular and hearing functions, which tells us ATP signaling is very important for inner ear to work properly. Through behavior tests and nerve signal measurements, it was found that P2X2 plays very important function for keeping balance sensory cells working properly, 37 and the KCNQ4 potassium channel not working well is a big reason why vestibular function goes wrong. Mice having Kcnq4 gene change shows bad hair cell electrical activity and cannot keep balance well, this means the channel is very important for inner ear ion balance, and this framework is a basic method to study potassium channel-related inherited balance problems, which was first proposed in previous research. Nowadays, more and more research has pay attention to the TRP (transient receptor potential) ion channel family’s important role in vestibular mechanisms, which is very crucial for understanding the inner ear function, for example, TRPV4 can be found in vestibular hair cells and also nerve tissues, if it is not there then may cause problems with balance. Mice without TRPV4 shows poor balance and movement control, which means this gene is important for feeling mechanical signals and cell communication, 38 and studies about ion channel models has found out the molecular reasons for vestibular disorders, also they pointed out good targets for gene and drug treatments. Gene replacement, genome editing, and drug control of ion channels shows good results, making progress in treating genetic vestibular diseases, and in the future, combining single-cell sequencing and high-resolution imaging can more clearly show how ion channels are expressed and controlled in different vestibular cells, which gives scientific support for precise treatments. With gene editing technology getting better, fixing ion channel genes inside living body may become a new way to treat inherited vestibular diseases.
Cytoskeletal protein–related mouse models of vestibular dysfunction
The cytoskeleton plays very important role in maintaining vestibular hair cells’ shape and sensing ability, also it helps organize stereocilia, transport materials inside cells and keep cell connections. Gene changes controlling cytoskeleton parts often cause stereocilia disorder and weaker mechanotransduction, which are main pathological features in genetic vestibular diseases. MYO6 is a myosin VI motor protein moving along actin filaments to minus-end direction, crucial for stereocilia structure stability. Mice with Myo6 gene mutation show serious hair cell stereocilia problems in inner ear, leading to balance issues, head tilting and circular movement. This model is commonly used to study how intracellular movement and stereocilia stability assist balance function. MYO7A, an unusual myosin type, has key function connecting stereocilia with cuticular plate and participating in mechanotransduction process. 39 Mice lacking Myo7a exhibit severe balance problems, frequently head shaking and abnormal swimming. This model is widely applied in Usher syndrome research and serves as typical structure for stereocilia development and maintenance study. 34 CDH23 and PCDH15 produce cadherin-like proteins located at stereocilia tip links of hair cells. 40 These linkings are very important because they can turn mechanical forces into electrical signals, which makes them very useful in many applications. If without them, the hearing process cannot function well. When these genes change, they harm tip-link growth leading to mechanotransduction failure and big troubles in vestibular function. Wong, E et al. found mice with Cdh23 and Pcdh15 mutations shows stereocilia adhesion issues can mess up balance signal transmission. ESPN (espin) is a protein that binds actin and very important for stereocilia to grow and keep. Mice lacking Espn has shorter and messy stereocilia, making their balance bad and posture unstable. This shows cytoskeletal connection how important for hair cell function. Studies about mouse models concerning cytoskeleton not just explain vestibular damage at cellular level but also point out possible gene therapy targets, like using AAV to correct MYO7A expression which was shown can recover balance in Myo7a-lacking mice. Furthermore, advanced HD imaging techniques such as super-resolution microscopy and cryo-electron tomography now allow clearly observing stereocilia’s complex structures, helping researchers better investigate cytoskeletal issues. Simply put, mouse models with cytoskeletal proteins plays key role in understanding how structural defects lead to vestibular disorders, while these structures also provide foundation for developing precise treatments aiming to restore cytoskeleton stability and hair cells’ mechanosensory function.
Mouse models of vestibular development–related genes
The vestibular system’s normal growth must have key genes working together, these genes control inner ear development, help neural circuits to form, and make balance function work well. Genetic diseases causing development problems is a major reason for inherited inner ear issues. The Pou3f4 (Brn4) gene is very important for otic capsule and semicircular canals development, because if missing, they cannot form correctly. This gene is very important because it helps the ear parts to grow in the right way. When Pou3f4 (Brn4) is not functioning good, the otic capsule and semicircular canals will develop problems. The mice without Pou3f4 gene has poor growth of auditory bony labyrinth, causing abnormal semicircular canals and bad vestibular function, which gives important real-world evidence for studying DFN3-related hearing loss genes and balance disorders together, just like Lanford and others 41 discovered. Fgfr3 gene is in charge of regulating the development of inner ear sensory cells and their supporting cells nearby, and mice missing Fgfr3 shows inner ear deformities and balance issues, proving its critical role in forming vestibular sensory cells and constructing neural networks. 42 The Notch pathway decides whether cells become auditory hair cells or supporting cells during vestibular development, and mutations in Notch-related genes results in incorrect vestibular system growth. 43 Slc17a6 gene produces VGLUT2, a vesicular glutamate transporter essential for vestibular synaptic transmission, and in mouse models mimicking spatial disorientation, Slc17a6 expression changes significantly, indicating its role in regulating vestibular nerve circuits and offering new insights for vestibular system research. 25 These gene models explain molecular mechanisms in vestibular organ formation and how developmental errors causes functional impairments, providing theory support for early detection and treatment of genetic vestibular diseases. Future studies combining genomic methods and single-cell technology will better understand gene networks in vestibular development, advancing personalized medicine.
Other important mouse models of vestibular dysfunction
Apart from these known models, other mouse systems born with balance problems also provide useful understandings about disease mechanisms. Mice without Cochlin develop worse balance issues when aging, similar to human DFNA9 genetic disease impacting inner ear, proving Cochlin is very crucial for vestibular system balance and extracellular matrix stability. 44 When Cochlin is insufficient, vestibular hair cells and neurons deteriorate causing balance dysfunction. Zpld1 mutant mice naturally develop semicircular canal defects, indicating this gene’s vital role in maintaining vestibular sensory organs’ structural integrity. Zpld1 mutations disrupt endolymph flow in semicircular canals, affecting vestibular signal processing and resulting in imbalance and movement disorders. 45 Studies on Sox10-deficient mice reveal neural crest cells’ importance for vestibular nerve formation, where insufficient Sox10 reduces vestibular ganglion cells and impairs function, linking poor neural crest development to inherited vestibular defects. 46 Importantly, several genes reported in mouse models of vestibular dysfunction have also been implicated in familial and sporadic Meniere disease, including FAM136A, DTNA, MYO7A, CDH23, PCDH15, and GJB2.47,48 Studies have identified an excess of rare missense variants in hearing loss genes in sporadic Meniere disease patients, while missense and loss-of-function variants contribute differently to the genetic structure of familial and sporadic forms.47,48 Furthermore, potential mouse models for Meniere disease such as those evaluated through rotarod and acoustic startle reflex performance have provided additional insights into vestibular dysfunction phenotypes relevant to this condition. 49 These models deepen our knowledge of genetic and mechanistic variations in hereditary balance disorders, laying experimental groundwork for targeted therapies. With advancing gene editing and multi-omics technologies, more hereditary vestibular disorder models are created, offering abundant resources to investigate disease mechanisms and therapeutic development.
Cross-species comparisons: Mouse and human vestibular genes
When translating findings from mouse models to human clinical applications, it is essential to consider both genetic homology and expression differences between species. BLAST analysis of candidate vestibular genes reveals high sequence conservation between mouse and human orthologs for most genes implicated in hereditary vestibular dysfunction. For example, MYO7A shares approximately 95% amino acid identity between mouse and human, while CDH23 and PCDH15 exhibit over 90% homology, supporting the translational validity of mouse models for studying Usher syndrome and related disorders. However, differences in gene expression patterns should also be considered, as methods described by Nadar-Ponniah et al. 30 have demonstrated that some genes show differential expression levels between mouse and human vestibular tissues, which may influence phenotype severity and disease progression. Comparative transcriptomic analyses further reveal that while core vestibular developmental pathways (FGF, Wnt, Notch, SHH) are conserved, temporal expression dynamics differ between species, with some human vestibular genes showing prolonged developmental expression windows compared to mice. These cross-species differences have important implications for interpreting mouse model data and designing therapeutic strategies, as treatment windows identified in mice may require adjustment for human patients.
Translational research progress: From mouse models to clinical application
Application of mouse models in elucidating pathological mechanisms
Family inherited inner ear diseases have many gene mutations and biochemical reasons behind. Mouse models with clear genetic background and easy gene editing is very good for studying disease mechanisms. Researchers using gene knockout, knock-in and natural mutation models can carefully observe changes in vestibular receptors, nerve pathways and brain processing, making studies more complete. The main causes of inherited balance problems are stereocilia growing wrong in hair cells, otolith organs not functioning well, and vestibular nerve signals getting confused. Though behavior signs not always show inner ear issues, but combining with tests like VsEP and VOR can accurately check vestibular function as Jones and Jones 23 reported. These systems has clearly shown the key role of mechanosensitive channels like TMC1/2, with mice missing Tmc1 showing bad balance problems, while Tmc2 knockout only cause lighter symptoms. 36 Also, structure protein changes such as Myo6 can make stereocilia abnormal and lead to balance trouble, proving cytoskeleton is very important for keeping hearing cells’ shape and work right. 50 Mouse experiments more clearly show how development genes like Pou3f4, Fgfr3, and Notch signaling parts function in vestibular system, and that embryo development problems can cause shape and function issues.41,42,51 By using behavior, tissue study, and molecular tests together, these models create strong system for researching inherited inner ear diseases’ causes and development. Recently, scientists using advanced imaging and gene editing methods copy human disease mutations in mice, then study their effects on vestibular system. These systems are useful tools for drug testing and gene therapy development, helping turn lab findings into real treatments. Combining molecular biology, genetics and neuroscience research improves understanding of hereditary vestibular disorders, providing basic ideas for new diagnosis and treatment ways. More studies now focus on how vestibular neurons worsen and adapt in hereditary balance disease. Research shows problems in axon transport and synapse dysfunction are very important for disrupting vestibular signal processing. 52 Mouse models help observe neuron structure and function changes, discovering neurodegeneration timelines and finding possible treatment targets.
Biomarker studies for diagnosis based on mouse models
In clinical practice, how to timely discover inherited balance disorders still faces many diagnosis problems that need solving. Mouse models is very useful for discovering and verifying diagnostic biomarkers, allowing researchers to investigate molecular changes and their connections with vestibular features at cellular and whole-body levels. Studies in Coch mutant mice found increased inflammatory mediators like IL-6 and ECM components such as fibronectin in vestibular tissues, which are related to tissue damage and poorer vestibular function, these substances can serve as markers for early diagnosis and treatment evaluation according to Chen, J. et al., 21 mouse model proteomics study found many secreted proteins in endolymph and perilymph changed a lot when vestibular degeneration become more serious. For example, BDNF and GDNF levels variation indicate hair cells and nerves damage, so they may serve as non-invasive markers for inner ear diseases, like Wang et al. reported, scientists also examined the inner ear fluids and tissues from genetically modified mice by applying advanced techniques such as transcriptomic and metabolic analysis, which discovered potential new biomarkers including oxidative stress-related metabolites and ion transport-associated mRNAs, helping to reveal disease mechanisms and offering diagnostic tools. Movement-based biomarkers like altered walking patterns, head tilting, swimming direction, and balance maintenance have been utilized in studies, providing additional methods to assess vestibular function, and when combined with molecular markers, they enhance diagnostic precision. 53 Animal studies allow continuous monitoring of biomarker changes, enabling tracking of disease progression and treatment efficacy, thus establishing a foundation for clinical molecular diagnostics, where accurate biomarkers can facilitate personalized diagnosis and timely intervention for genetic balance disorders, which is of great significance. 54
Application of mouse models in the development of therapeutic strategies
The progress in treating genetic ear diseases heavily depends on reliable mouse models that plays a key role. Significant advances has been achieved in gene therapy, genome editing and drug treatments through mouse models, which lays the groundwork for disease cure. In gene therapy research, using AAV to deliver genes showed good effects in Vglut3-deficient mice, improving their hearing, similar approaches were also tested for balance disorder treatment. This study not only confirms gene replacement therapy’s effectiveness but also emphasizes the critical timing of treatment, proving early intervention is necessary for functional recovery. 55 By applying ASO targeting USH1C mutations, hearing and balance functions was greatly improved in genetically modified mice, with the optimal treatment window identified as immediately after birth. These findings demonstrate timely treatment’s importance and provide solid evidence for genetic approaches to fix inherited balance issues. 27 The CRISPR/Cas9 gene editing technology is increasingly adopted in mouse experimental models. For instance, CRISPR correction of Tmc1 defects successfully restored hearing and balance capabilities, indicating genetic modification’s great therapeutic potential. This method holds big advantages because it can accurately repair disease-causing genetic errors while causing fewer unintended changes, making it highly promising for therapy. 56 Regarding drug treatments, α1-antitrypsin (AAT) demonstrated superior recovery effects compared to traditional corticosteroid therapy in vestibular-damaged mice. Research reveals AAT improves vestibular function by combating inflammation and protecting nerves, offering clinicians new treatment options. 57 Furthermore, hormone regulators like thyroid hormone aids vestibular recovery and enhances balance ability. Thyroid hormones facilitate neural adaptation and energy regulation, supporting ear balance restoration and showing therapeutic value. 58 Rodent models serve crucial role in testing new drugs and treatment methods’ efficacy and safety for medical research. Through behavioral tests, electrophysiological recordings and histological examinations, researchers can comprehensively assess treatment outcomes and refine therapeutic approaches. Future continuous improvements in gene editing tools and nano-delivery systems will further elevate mouse models’ importance for precise inherited inner ear disease treatment. Integrating multi-omics and systems biology enables personalized therapies while facilitating long-term patient monitoring.
Current status of clinical translational research
Mouse model studies are gradually moving towards hospital use, but many big problems still waiting to be solved. Due to the inner ear’s complex structure and sensitive working mechanism, drug delivery systems must consider not only effectiveness but also body compatibility issues. Although adeno-associated virus (AAV) as gene delivery tool can successfully transfer genes to inner ear cells via round window membrane method, problems like low transfection efficiency and immune response remains to be improved, as many studies including Pan et al.
28
have shown that enhancing delivery methods is crucial for clinical gene therapy success, and because treatment window is limited, early intervention can achieve optimal results. In clinical diagnosis and treatment, the timing must be very precise and plays a crucial role. This is the reason why choosing right time must be paid special attention. For example, gene therapy works best in early babyhood but shows less effect in later stages, so quick diagnosis is very important for good results as Emptoz, A et al.
27
said. Now clinical trials are using both gene therapy and genome-editing methods, and studies about genetic hearing problems has started with balance ability recovery seen as key measure standard. Scientists research AAV gene therapy progress for inner ear problems, saying vestibular function tests is very important in clinic checks according to Stone et al.
46
Because inherited vestibular diseases have many types and patients are different, treatment results can vary much so accurate patient classification and personalized treatment plans are highly needed. By combining genomic data, high-tech imaging, and body function tests, doctors can make personalized plans to improve outcomes as Maudoux et al.
58
suggested. Diagnostic tools make progress for clinical use, where advanced MRI and vHIT test can better diagnose vestibular problems, helping find early signs and monitor treatment progress. Moreover, 3D imaging for endolymphatic hydrops gives new ways to detect sickness like Menière’s disease according to Noh et al. Going forward, cross-field cooperation and big data analysis will push genetic vestibular disorder research into clinical use. Mouse models keep improving and can nearly copy human diseases, making strong foundation for treatment success. With genetic modification and nanoscience making big progress, developing targeted therapies for ear diseases now looks more hopeful (Figure 2). Applications and challenges of HVDs mouse models in clinical translation.
Conclusion
Hereditary vestibular dysfunction is a complex inner ear disease caused by many different genes and showing various clinical symptoms. Mouse models, due to their clear genetic background and easy manipulation, have become crucial tools for researching disease mechanisms, discovering biomarkers, and developing treatments. Models for Usher syndrome, ion channel disorders, cytoskeletal and developmental gene mutations have successfully reproduced the symptoms and functional impairments of inherited inner ear diseases, providing powerful tools for both basic research and clinical applications. These models can not only reveal cellular and molecular mechanisms of disease progression, but also enables early detection and precise treatment through behavioral tests, electrophysiological signals, and microscopic analysis. Recent advances in gene therapy and gene editing, together with mouse model-based drug development, brings new hope for clinical translation. Techniques like adeno-associated virus gene delivery, CRISPR/Cas9 genome correction, and neuroprotective drugs administration all demonstrated good results in mouse models. However, clinical application still faces major challenges including improving delivery system efficiency and safety, optimizing therapeutic windows, considering individual differences, and creating personalized therapies. Future research should combine genomics, transcriptomics with single-cell technologies to better understand disease causes, while employing advanced imaging and diagnostic methods to enhance clinical accuracy. In conclusion, mouse models play vital role in bridging basic research and medical practice for genetic inner ear disorders. With continuous biotechnology progress and interdisciplinary collaboration, these diseases may soon achieve accurate diagnosis and effective treatment in near future.
Footnotes
Acknowledgments
I would like to thank the members of Heping Hospital Affiliated to Changzhi Medical College and Second hospital of Shanxi Medical University who have provided me with comprehensive professional instruction and guided me in scientific research.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province (20250053).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The authors have nothing to report.