
Abstract
In this paper we discuss two universal characteristics of domesticated species that distinguish them from the wild closely related ancestors – increased socialization and phenotypic variability. Examining evidence accumulated in the literature up to date, we note that the gut microbiome is involved in the increased social behavior of domesticated species through the gut-immune system-brain axis. We further discuss data that point toward clear difference in the microbiome composition between domesticated species and closely related wild ancestors. This difference is related to changes in diet, due to co-habitation with humans, which leads to increase in Bifidobacteria and changes in carbohydrate metabolism. We note that these changes may also influence interaction between microbiome and virome. Virome is linked to the evolutionary changes through incorporation of retro-viruses into the host genome. Together with transposons these mobile genetic elements may also lead to changes in regulatory networks, and increase adaptive potential. Changes in microbiome of animals during co-habitation with humans should be considered as an important event during domestication process.
C “All disease begins in the gut.” –Hippocrates of Kos (Hippokráte s ho Kṓos: c. 460–c. 370)
In a long-term perspective, domestication is a formation of a common niche between human and animals, resulting in many changes at systems levels for all participants (Zeder, 2020). Supposedly, “Domestication syndrome,” a complex of phenotypic, genotypic, molecular, and behavior traits, is shared by all of the domesticated species. Discussions about similarities of domesticated species, their differences from closely related wild ancestors, as well as whether human species can be considered domesticated, all started many centuries ago (Del Savio and Mameli, 2020). Although domestication process started relatively recently (Figure 1), it is an example of evolutionary process (Wilkins, 2020; Zeder, 2018).
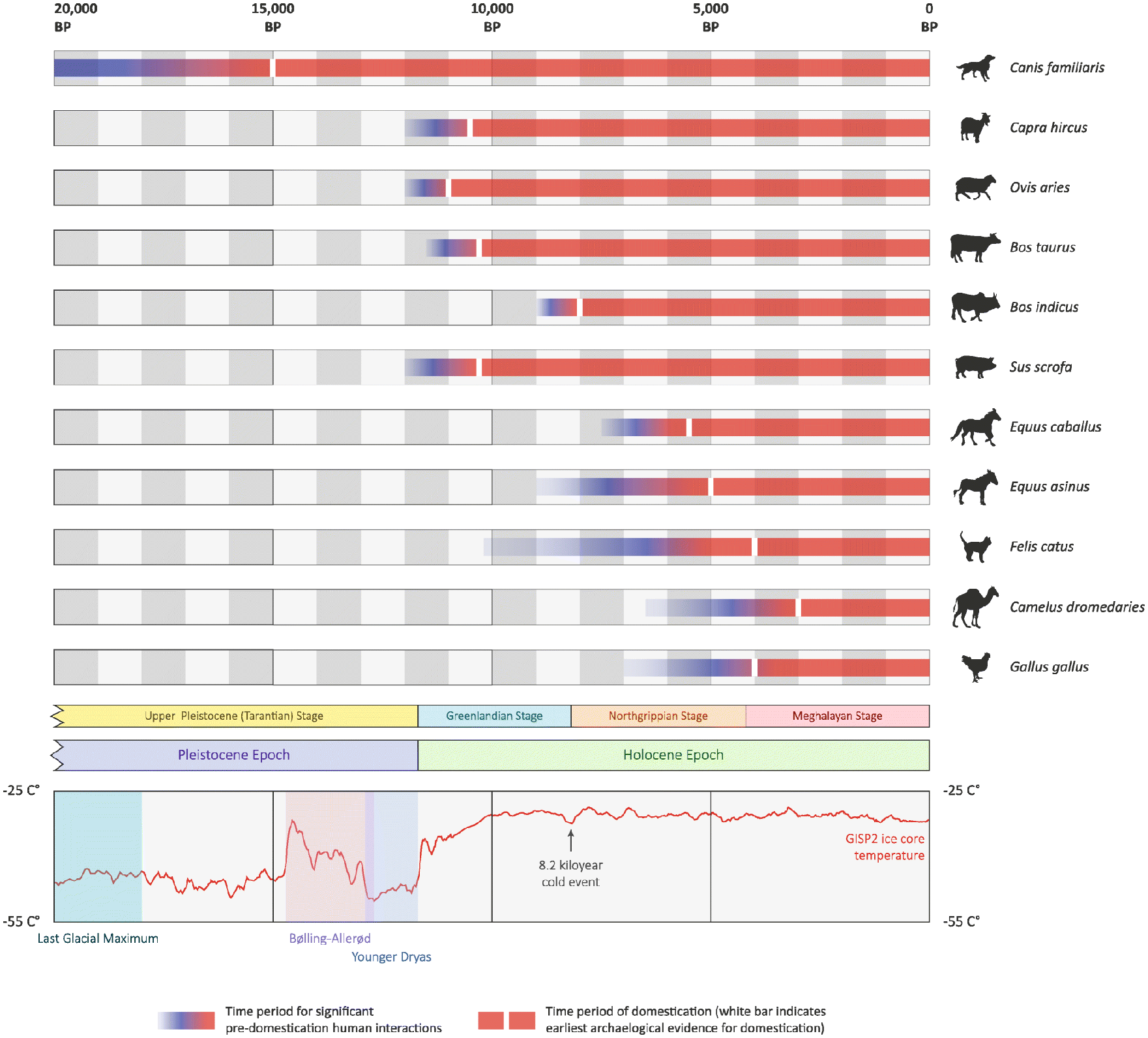
Timelines of domestication for 11 animal species with relevant stratigraphy and climate chronologies. For each species, the time periods of significant pre-domestication human–animal interactions are also shown (McHugo et al., 2019). Domestication timeline data (Larson and Fuller, 2015; Larson et al., 2014; Vigne 2015).
However, thus far even the traits defining domestication syndrome remain controversial (Sanchez-Villagra and van Schaik, 2019; Zeder, 2020). Different species are significantly different in what concerns interaction with humans, involvement in artificial selection, and benefits of their employment in a commonly created niche. The traits, defining domestication syndrome, also vary. Most frequently they include docility and tameness, coat color changes, reduction in tooth size, changes in craniofacial morphology, decrease in sensitivity of auditory, vision and olfactory abilities, reduction in total brain size, neoteny (accelerated sexual maturity), decrease in sexual dimorphism (feminization), more frequent estrus cycles, and several others. From literature about domestication traits, one can see that in general inter-species differences in manifesting domestication syndrome allow estimating its variability as purely quantitative trait. Some species have one or two traits that are different between domesticated and closely related wild animals, like for example between Bubalus and Yak, or all seven like in Canis familiaris (Sanchez-Villagra and van Schaik, 2019; Zeder, 2020). Though, the universal characteristic of domesticated species is a high level of phenotypic and population genetic variability that is not explained by interspecies hybridization, sometimes observed in wild animals (Figure 2).

Evolution and phenotypic diversity of domestic animals. The wild progenitor species are shown on the left and the domesticated animals are shown on the right. Except for the aurochs, all wild progenitor species are extant.
Recently, comparative genomics of dogs, yaks, poultry, rice and soybeans, and their wild closely related species demonstrated that domesticated species have high frequency of deleterious genetic variation (Makino et al., 2018). It was suggested that accumulation of these variants started relatively recently and was caused by artificial selection approaches that were getting more and more sophisticated with time. It was also found that regulatory sequences contribute significantly in genomic variability (Kingsley et al., 2019).
To summarize, current data suggest that the most universal domestication traits are behavioral changes (docility and tameness) (Wilkins, 2020) and significant increase in phenotypic and population genetic variability (Glazko et al., 2015). Then, the most important question in finding domestication mechanisms is how exactly the variability of these two intrinsically different universal domestication traits is related.
Microbiome and behavioral traits
One of the mechanisms influencing behavioral traits could be changes in animal’s microbiome, caused by a specific niche formed by cohabitation with humans. Gut microbiome was recently called a “forgotten organ” (O’Hara and Shanahan, 2006) and became a focus of new theory – microbial involvement in evolutionary processes (Kolodny et al., 2020). All species have species-specific gut microbiome, influencing their adaptation and diversification, helping with challenges of food availability, phenotypic plasticity, innate, and adaptive immunity. Generally, gut microbiome is an important environmental factor and selective force, forming adaptive evolution of animal’s diet, phenotypic plasticity, gut morphology, and immunity (Kolodny et al., 2020).
The theory, studying the role of interactions between macro and microorganisms during evolution is now called the “hologenome theory of evolution.” The union of all the genes in the holobiont, that is all the genes in the microbiome plus the genes of the host, constitutes the hologenome (Zilber-Rosenberg and Rosenberg, 2008). The hologenome theory considers interactions in hologenome as a main focus of genomic changes under environmental pressure (Zilber-Rosenberg and Rosenberg, 2008).
Different environmental microbial species colonize newborn intestinal tract after birth and form gut microbiome, a complex system with more cells than there are somatic cells in human organism and approximately 150 times more genes than there is in human genome. Recent data suggest that gut microbiome influence regulation and development of hypothalamic–pituitary–adrenal axis, controlling many behavioral reactions as well as digestive and immune systems. It is assumed that interactions between host and microbiome occur through different receptor molecules in the intestinal tract (Douglas-Escobar et al., 2013). The microbiome-gut-brain axe and its variability influence cognition and behavior (Petra et al., 2015; Rieder et al., 2017).
Accumulated evidence suggests that brain development and physiology are influenced by gut microbiome. Gut microbiome can send signals to brain through many different ways, including microbial peptides and metabolites, immune activity, activation of nervus vagus, and neuromodulators production. As a whole this two-ways system is known as microbiome-gut-brain axe. Germ-free mice treated with antibiotics had shown changes in many central physiological processes, such as neuromodulators turnover, neuroinflammation, neurogenesis, and neurons’ morphology. Probably this is why germ-free mice behavior is radically different from social behavior of conventional mice. Interestingly, supplementing germ-free mice with probiotic bacterial species (e.g. Bifidobacterium and Lactobacillus) improves social behavior. These results underscore the importance of microbiome signaling for central nervous system development and social behavior programming (Cryan et al., 2019). The studies of host-microbiome interactions are ongoing, however the question why and when the gut-brain interactions, defining social behavior, appeared during the evolutionary process, still remains unanswered (Sherwin et al., 2019). The multitude of research currently exploring the microbiome-gut-brain axe in various diseases, including desocialization, allows expecting the development of new treatment methods employing bacteria (Cryan et al., 2019). Accumulated data suggest that people with more diverse social contacts, as a rule, have more diverse microbiome than people with less social contacts. Probably social interactions have some impact on forming gut microbiome. In contrast, anxiety and stress lead to decrease in microbiome diversity and changes in its bacterial content (Johnson, 2020).
It is generally believed that central nervous system (CNS) is an important regulator of intestinal health and CNS dysfunction may lead to quick intestinal health deterioration. Patients with neuroimmunological and neuroinflammatory intestinal health symptoms are not well monitored, but intestinal health is vital for their life quality. Contemporary treatment strategies frequently applied by gastroenterologist and neurologist separately raise a question if the treatments are as beneficial as they could be if applied by two specialists in concordance, consulting with each other. Neuro-immune gastroenterology is an area of research in between several medical sub-specialties. Despite the accumulated data about neurological disorders that are influenced by immune system, and their involvement into intestinal health, neuro-immune gastroenterology still does not influence neurological treatment plans (Figure 3).
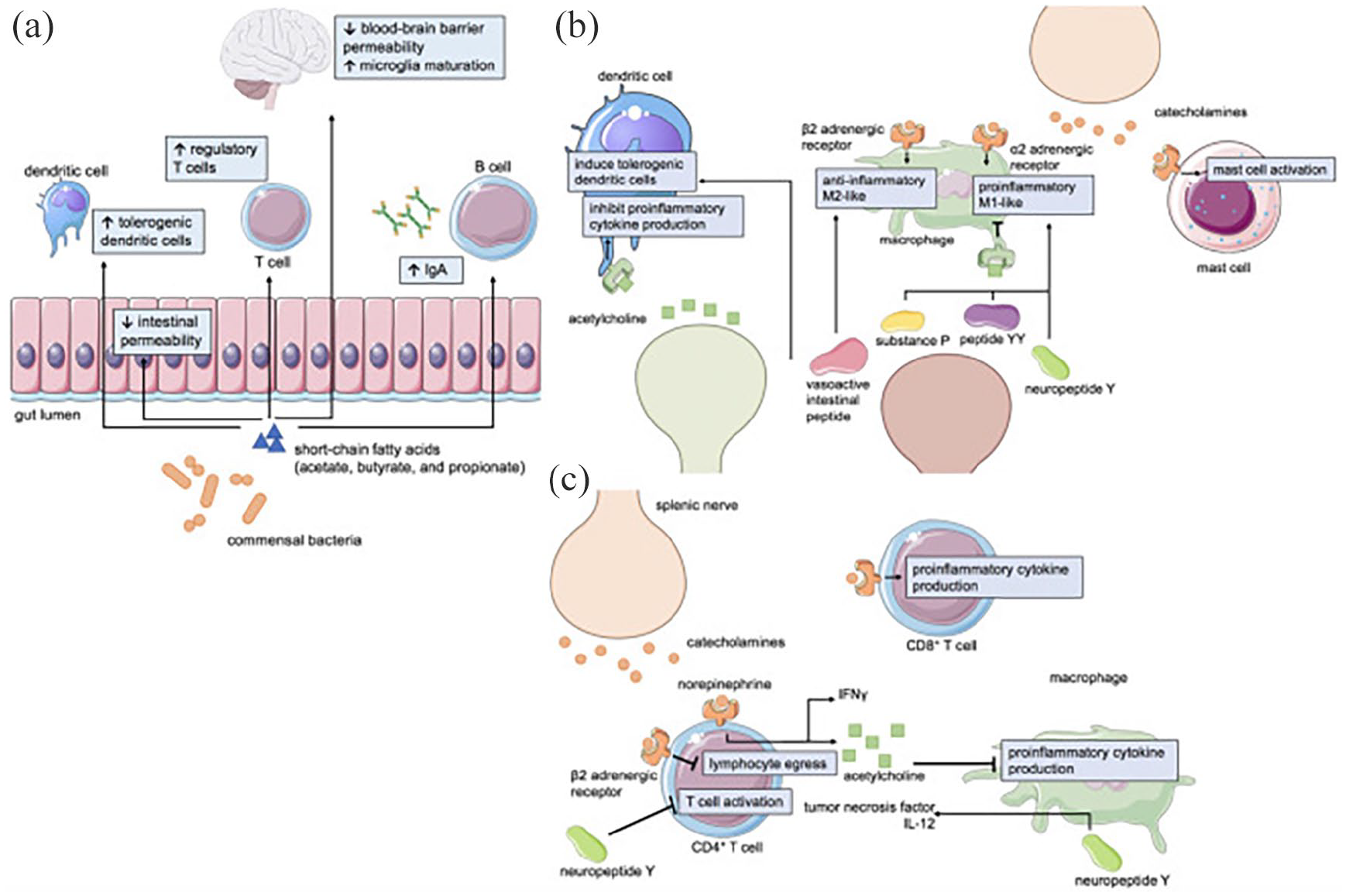
Mechanisms of neuroimmune-gastrointestinal crosstalk. (a) Short-chain fatty acids (SCFAs) such as acetate, butyrate, and propionate are metabolites generated by commensal gut bacteria that affect intestinal permeability, immunity, and CNS physiology. Neurotransmitters and neuropeptides can influence the function of
Microbiome and virome
Right now many studies are focused on showing how microbiome may protect host from viral infections, in particularly COVID-19 (Figures 4 and 5) (Antunes et al., 2020), and how it can ameliorate pathogen-induced gut dysbiosis (Trottein and Sokol, 2020).

Dysbiosis process, a perturbation of the microbial community, it may decrease the microbiota diversity, shift its composition and, consequently, facilitate the invasion and viral replication. It might create an inflammatory environment explored by SARS-CoV-2. Pathogens microbes and virus contribute toward an intestinal and lung barrier dysfunction, with liberations of pro-inflammatory cytokines, promoting a “cytokine storm.” Unhealthy diet might reduce the phagocytic activity and modulation of the production of immunoglobulins/antibodies that mediate host defense, lowering the elimination intracellular pathogens and the immune response.

Main mechanisms of action of fermented foods, probiotics, and prebiotics. Prebiotics are selectively utilized by the commensal microbiota, releasing metabolites like short chain fatty acids (SCFA), promoting leukocyte recruitment to the site of infection, as well as their activation. The SARS-CoV-2 binds to the ACE2. Fermented foods and probiotics strains may also increase the phagocytic activity and modulate the production of immunoglobulins/antibodies mediate host defense by eliminating intracellular pathogens, improving the immune response, intestinal microbiota has a marked influence on metabolic pathways within alveolar macrophages, which correlates with an altered cellular responsiveness, promoting the microbiota modulation has a marked influence on metabolic pathways within alveolar macrophages, nutritional interventions of omega-3 polyunsaturated fatty acids, selenium, zinc, iron, vitamins A, B2, B3, B6, C, D, and E to combat viral infections. ACE2, angiotensin-converting enzyme 2; L-cell, enteroendocrine L-cell; M cell, microfold cell; SARS-CoV-2, severe acute respiratory syndrome.
Some bacterial products influence host immune response during respiratory viral infections. They can also regulate systemic inflammation and endothelial damage, two biggest concerns with COVID-19 infection (Figure 6) (Infusino et al., 2020).

Hypothesis on the mechanism of endothelial involvement (a) and intestinal involvement (b) and in coronavirus disease.
Right now accumulated data suggest that bacterial therapy can, potentially, be used in SARS-CoV-2 treatment. A group of patients infected with SARS-CoV-2, matched by age, sex, background pathology, lab results, and oxygenation were separated into two groups, one getting bacterial therapy and another one “control” group. During the next 72 h in the group receiving bacterial therapy remission of diarrhea and other symptoms was observed, as compared with the less than a half of the other group. Expected risk of developing severe respiratory symptoms was less than eight times in patients receiving per-oral bacterial therapy. The severity of the disease as well as the death rate of patients in the intensive care unit, were higher in patients not receiving per-oral bacterial therapy. The authors conclude that the specific bacterial composition they were using for therapy was providing an improvement in clinical characteristics of patients, infected with COVID-19. The results also point toward an importance of gut-lungs axis in COVID-19 (d’Ettorre et al., 2020).
It is already well known that the susceptibility to infections and disease outcomes in humans and farm animals are influenced by microbiome (Zeder, 2018). In some studies it was shown that microbiome directly influence the host susceptibility to pathogens. Recently, bacterial communities were obtained from feces of domestic pigs (Sus scrofa) rose under different conditions: specific-pathogen-free (SPF) pigs and domestic pigs from the same bred, and indigenous domestic pigs from a backyard farm in Kenya. Also, the fecal microbiota composition of the African swine fever (ASF) resistant warthogs (Phacochoerus africanus) from Africa and a European zoo was obtained. African swine fever (ASF) is a devastating disease for domestic pigs. African animals showed the highest microbial diversity while the SPF pigs the lowest. Analysis of the core microbiota from warthogs (resistant to ASF) and pigs (susceptible to ASF) showed 45 shared OTUs, while 6 OTUs were exclusively present in resistant animals. These six OTUs were members of the Moraxellaceae family, Pseudomonadales order and Paludibacter, Anaeroplasma, Petrimonas, and Moraxella genera (Correa-Fiz et al., 2019).
Among various host pathogenic infections, the most serious problem in health care world-wide present viral infections. Commensal microbiota interacts closely with viruses during infection process. Accumulated data suggest that commensal microbiota regulates, and in turn is regulated by viruses through different mechanisms, playing either stimulating or suppressing role in viral infections (Figures 7 and 8). In addition, the integrity of commensal microbiota is changing with viral intervention resulting in host’ dysbiosis and influencing host’ viral resistance (Li et al., 2019).
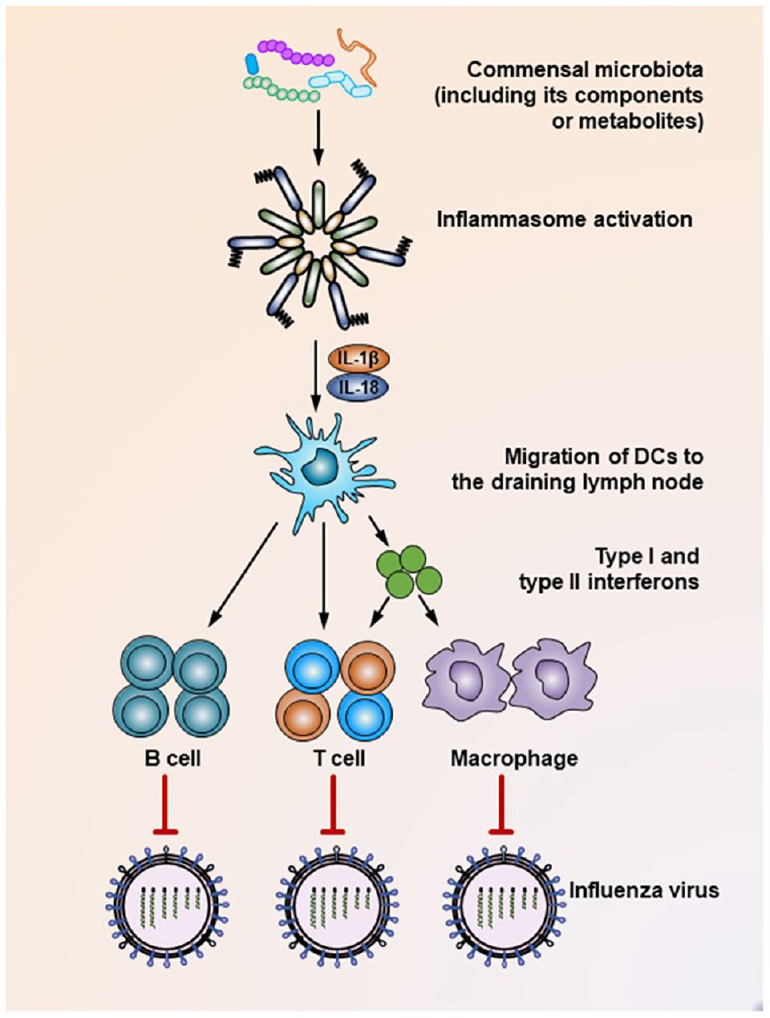
Mechanisms underlying the suppression of influenza virus infection by the commensal microbiota. During the influenza virus infection, organisms of the commensal microbiota, as well as their components (i.e. various TLR ligands) or metabolites (i.e. desaminotyrosine) activate the inflammasome, resulting in IL-1β and IL-18 production. The production of these two cytokines induces the migration of dendritic cells from the lung to the draining lymph nodes, where they act as antigen-presenting cells to prime virus-specific B cells, CD4+ T cells, CD8+ T cells, and macrophages. In addition, dendritic cells also secrete type I and type II interferons to stimulate the activation of T cells or macrophages. As a result, these effector cells secrete virus-specific antibodies or inflammatory cytokines or exert direct virus-killing effects to suppress the infection process of the influenza virus.
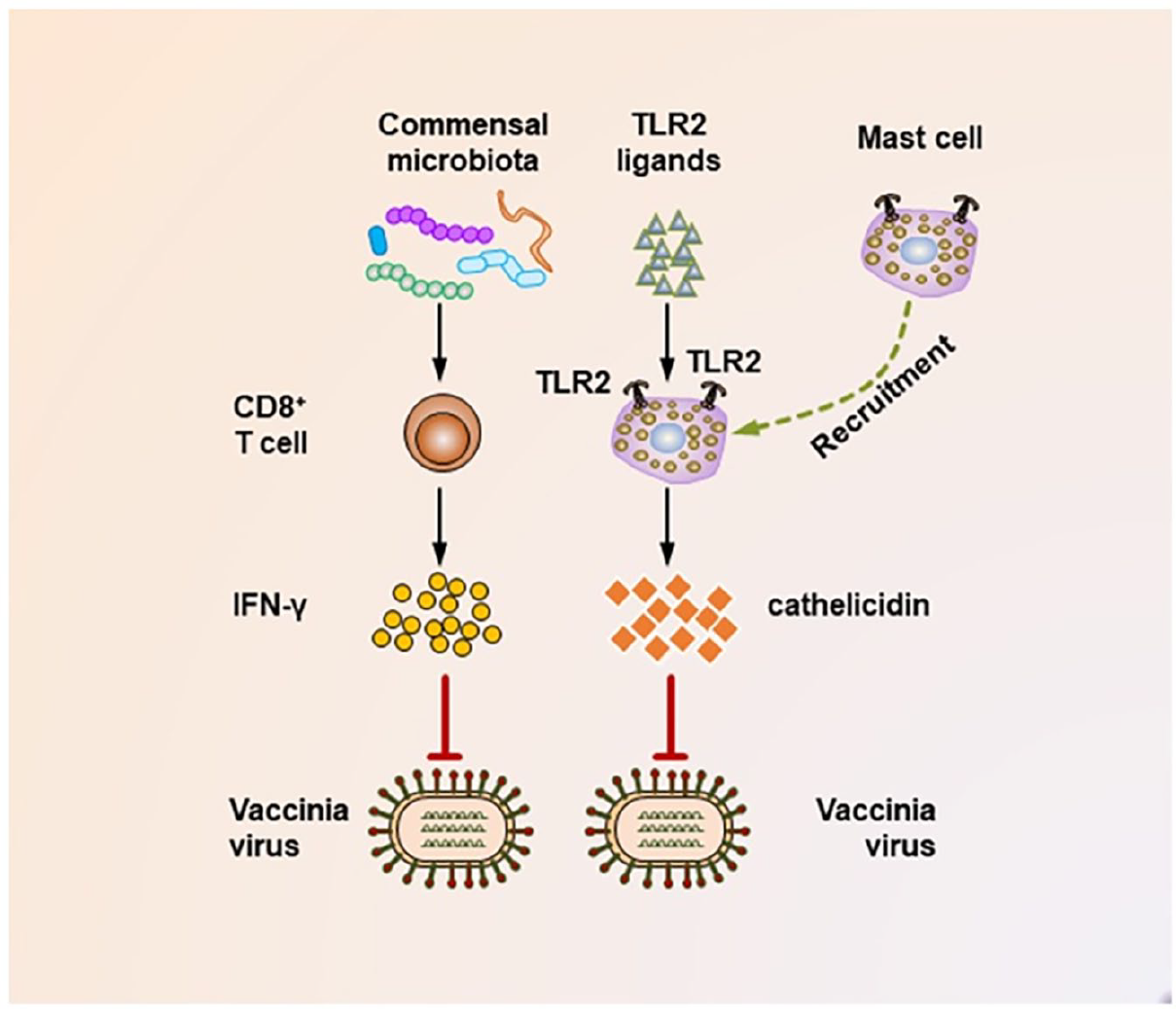
Mechanisms underlying the suppression of vaccinia virus infection by the commensal microbiota. During the vaccinia virus infection, the commensal microbiota primes virus-specific CD8+ T cells to secrete large amounts of IFN-γ, which critically mediates the corresponding antiviral immunity. In addition, during vaccinia virus infections, the activation of TLR2 by bacterial products is essential for recruiting mast cells to sites of viral infection. These mast cells also contribute to suppressing the viral infection by secreting an antiviral cathelicidin.
It should be noted that host-microbiome-virom interactions are complex and include many steps, in particular host’s microbial resistance and microbiome’s viral resistance. The last decades had shown that “repressive” medicine, namely the one preferably using antibiotics, was not very effective in veterinary, because repetitive use of antibiotics is quickly causing emerging microbial resistance. Microbial antibiotic resistance required the development of new tools protecting humans and farm animals against infectious diseases. In 2017 EU accepted “united health” approach to prevent antibiotic microbial resistance in human and animals, as well as transmission of zoonotic diseases. Bacteriophages (viruses that infect bacteria) demonstrate antimicrobial activity toward a wide spectrum of effective and specific pathogens. Bacteriophages are crucial for microbial evolution and are important parts of many ecosystems, including human microbiome. Their specific properties, such as antimicrobial activity, specificity, and transmissibility make them important therapeutic agents (Garvey, 2020).
Microbiome and farm animals
It was already mentioned that microbiome can play a key role in formation of gut-brain axe. There are five observations, supporting this hypothesis: in the absence of microbiome there is severe brain damage; feeding with specific bacteria lead to behavioral changes in animals; population research of people infected with specific pathogens demonstrate symptomatic changes in brain-gut diseases; in different age groups antibiotic treatment influenced brain, spinal cord, and gut-brain connection; some infectious diseases can be treated with microbiome (Cryan et al., 2019). These observations led to the idea of existence microbiome gut-brain axe.
One of the most important research directions in studying microbiome-gut-brain axe is research and analysis of the accumulated data that gut microbiome influence nervous system development and social behavior in different animals. This tight interconnection between host and microbiome hints that the interactions between host and microbiome were influencing the evolution of social behavior. Indeed, some species use gut microbiome to facilitate social interactions (Sherwin et al., 2019).
The wide spectrum of social behavior, including aggression, depression, and anxiety is defined by interactions between many physiological systems, including neuroendocrine and immune systems. Gut microbiome plays an important role in social modulation and probably serves as an important integrator of other physiological systems. Microbes constituting gut microbiome can interact with brain axe using neural and humoral pathways. There is a highly complex intricate relationship between brain, microbiome and immune system, and their ability to influence behavior (Sylvia and Demas, 2018). In this context the question about the differences between gut microbiome in domesticated and closely related wild animals’ is intriguing and of high priority (Alessandri et al., 2019).
All multicellular organisms benefit from their microbiomes that play a central role in host gut health and immunity. Recently there was an exponential growth in researches concerning microbiomes of economically important animals, fishes, and plants, because it is assumed that productivity and resistance of farm species can be improved by manipulating of microbiome. However, microbiome optimization remains a challenging task because there is not enough knowledge about dominant host’ microorganisms or substantial differences in hosts microbiomes. In order to gain more knowledge about what could be an “optimized microbiome” the data about microbiomes in farm animals, fishes, and plants were summarized (Figure 9). The data emphases bacterial communities structure, their metabolic functions, as well as how microbiome can be influenced by domestication, and traditional agricultural practices using antibiotics (Ikeda-Ohtsubo et al., 2018).

Microbiota in agriculture. The figure provides an overview of the bacterial composition of the microbiota of different parts of livestock animals, gill and intestines of fish, and phyllosphere and rhizosphere of plants at the phylum-level (pie-charts) and lower taxonomic levels. The data sources are 16S rRNA or metagenomic analyses.
High prevalence of Proteobacteria in animals, fishes, and plants (Figure 9) reflects the benefits facultative anaerobes have in host’ milieu, where strict anaerobes risk having toxic oxygen and strict aerobes risk fighting with the other bacteria for oxygen as an electron acceptor. The oxygen-anoxic background is a typical environment in host milieu and is a major factor in determining host microbiome. Facultative anaerobes have highly flexible metabolism: they can produce energy by fermentation, use inorganic nitrogen compounds, for example nitrate as an alternative electron acceptor toward oxygen. Given oxygen they grow fast using oxygen, providing and destroying various organic compounds that change significantly the background of their environment. The exceptional adaptability of Proteobacteria to constantly changing environment assume high underlying phenotypic and genotypic plasticity and allows Proteobacteria to be generalists in host associations, mostly presented by target symbionts and pathogens (Ikeda-Ohtsubo et al., 2018).
Microbiome investigations of farm animals show how domestication process was influencing microbiome content of agricultural species. Comparative analysis of gut microbiomes in pigs and wild boars has shown that Lactobacillus spp., dominating pigs gut microbiome, were absent in wild boars. Interestingly, dominating species in microbiome’s of recently domesticated wild boars were Enterobacteriaceae, suggesting that gut microbiome of domesticated pigs reflects agricultural management. It is well known that the abundance of Enterobacteriaceae correlates with diarrhea after weaning, therefore the pigs’ ration can significantly influence their health through variations in gut microbiome. It was also shown that cattle inoculated with the content of bison’s tripe had increases ability to digest proteins and higher content of nitrogen, but not cellulose digestion. Presumably the microbiomes of contemporary domesticated species ancestors were better able to extract nitrogen from roughage (Ikeda-Ohtsubo et al., 2018).
Comparative analyses of zebrafish microbiomes in laboratory animals and free living zebrafishes produced controversial results. Several studies did not find any differences, while some others did show the significant decrease in microbial diversity for laboratory populations (Ikeda-Ohtsubo et al., 2018).
Plant microbiome is also significantly influenced by domestication and selection process. Artificial selection toward increased productivity of economically important plants and the use of fertilizers and pesticides result in selection of specific plants traits based on the maximal use of roots microbiomes. Various researches found very specific components of plants microbiomes of domesticated and closely related wild plants, for example rhizospheric microbiome component root beets and endophytic component in grape vine. Interestingly, plant hosts react to microbiome changes by changing their physiology and in this way manipulate their microbiomes better than animals (Ikeda-Ohtsubo et al., 2018).
Domestication is the process when humans change life and behavior of wild animals in the way that fits better human needs. To understand how domestication influences gut microbiome of animals and identify specific bacterial populations, 16S rRNA sequencing and analysis were implemented on 146 fecal samples of domesticated and closely related wild animals pairs. It was shown that domestication led to more effective food into body mass transformation and presumably horizontal transfer of specific bacteria from human to domesticated animals, as compared to their closely related wild ancestors. Also it was found that fecal microbiome communities were adapting to specific food, that was shown as an increased abundance of glycoside hydrolases, probably because of higher consumption of complex plants carbohydrates (Alessandri et al., 2019; Larson et al., 2014).
Especially interesting are Bifidobacteria’s distribution and abundance. Comparative microbiome analysis for the presence of Bifidobacteria was done for hares, wolfs, boars, and their closely related domesticated species, namely rabbits, dogs, and pigs, respectively. Bifidobacteria diversity was higher in all domesticated species as compared to their closely related wild animals. It confirms that frequent interactions with human and domesticated lifestyle in those mammals lead to acquisition of additional Bifidobacteria taxa, because Bifidobacteria and human share the same niche. Also, the influence of artificial selection on gut microbial communities was recently studied. Feces samples were taken from wolf and seven breeds of dogs living in the same resort. The data suggest that bifidobacterial profiles are in agreement with phylogenetic relationships between species as well as between breeds. Thus, domestication and artificial selection of wild wolf gradually formed microbiome of mammal Canis lupus. In the process of domestication gut microbiome composition was presumable influenced by contacts with humans (Milani et al., 2017).
Virome and regulatory networks in domestication process
Recent viral metagenomics studies have shown that there is a wide variety of eukaryotic viruses and phages in feces of domesticated animals. For example, in healthy commercial minks from 40 farms of South Ontario plenty of phage sequences were found, mostly from the families Siphoviridae (53%), Podoviridae (22%), Myoviridae (20%), Inoviridae (1%), Leviviridae (0,04%), Tectiviridae (0,01%), and Microviridae (0.01%). A large spectrum of Vertebrate viruses was also found, where the most abundant were Posavirus 3, Mink Bocavirus, gyroviruses, and some bird viruses. In addition, Invertebrate viruses, associated with amoebas and seaweeds. Those results demonstrate that viral populations in healthy mink feces are diverse, abundant, and could be related with animals diet (Xie et al., 2019).
The accumulated evidence suggests that symbiotic relationships between host and microbiome form the axe: gut microbiome – immune system – brain, significantly influencing animals’ behavior and the ability to socialize. This axe naturally contains a “sub-axe” – virome – microbiome – immune system. Whole genome sequencing methods convincingly demonstrated that virome – microbiome – immune system sub-axe is actually virome – microbiome – immune system – genome sub-axe. Approximately half of all mammalian genomes is represented by viral and pro-virile sequences and their recombinant products, that form mobilome, – a mixture of mobile genetic elements or transposons.
It is generally believed that in mammalian genomes the activation of transposable elements (TE) lead to genomic instability, promote aging, and inflammation. Unlike mammalian cells, prokaryotes are constantly exchanging their genetic materials using TE, crossing interspecies barriers and facilitating fast microbial evolution, and microbial diversity in complex environments, such as mammalian gut microbiome. In microbiome, TE can escape prokaryotic cells or pathogens and cross the barrier between prokaryotic and eukaryotic cells. Supposedly this horizontal TE transfer from symbiotic microbes to eukaryotic host cells is a random process however constantly catalyzing genomic instability. The instability in turn results in structural and epigenetic variations of host genome as well as in activation of host defense mechanism that can promote biological aging and inflammation. Some accumulated evidence suggest that some steps in the host innate immunity can protect host from TE horizontal gene transfer (Figure 10, and also see Table 1 in reference 36 Greenwood et al., 2018) (Chalmers and Wu, 2020; Greenwood et al., 2018).

(a) An integrated double-stranded DNA provirus (yellow) of a simple orthoretrovirus within the host genome (gray) is shown. The long terminal repeats (LTRs) are at both the 5′ and 3′ ends of the provirus and flank the retroviral gag, pol, and env coding regions. Regions coding for enzymes and other proteins are shown with font colors corresponding to their depiction in panel B. (b) Schematic drawing of a simple orthoretrovirus. All orthoretroviruses have three component parts: (i) the RNA genome, shown in yellow; (ii) internal proteins, shown in red, including internal structural proteins (Gag) as well as the viral enzymes, including the reverse transcriptase (Pol), which makes a DNA copy of the RNA genome which will be integrated into the host cell genome; and (iii) the envelope proteins, shown in blue. Env consists of two components: the TM moiety is embedded in the membrane (depicted in gray and white) of a host cell and is incorporated into the virion during the budding process, and the surface glycoprotein SU forms the knobs and is the part of the virus that binds to receptors on susceptible cells of the host.
There is accumulated evidence that endogenous retroviral genes that are not expressed under normal circumstances, in some cases get expressed and translated. These cases become the basis for behavioral changes and their pathology (Johansson et al., 2020).
TE ability to move around genome provides them with opportunities to participate in large scale genomic rearrangements, with some of them being beneficial for the host, leading to TE long term preservation in the host genome. Frequently, TEs bear transcription factor (TF) binding sites and acquire the new ones during their transposition, providing random TFs insertions for the entire host genome. In this way TEs stimulate the evolution and diversification of gene regulatory networks and supply targets to regulate genes transcription. This phenomenon can explain fast and frequent evolution of new phenotypic features – for example convergent evolution of embryo nourishing tissues, the placenta in mammals, and the endosperm in flowering plants (Qiu and Kohler, 2020).
The analysis of TE genomic distribution demonstrates that mammalian genomes bear hundred thousand endogenic retroviruses acquired via ancient retroviral infections. It was shown that there is an intercontinental distribution of the retroviruses, the interspecies transmissions in at least 11 mammalian species and that recombination plays an important role in viral diversification (Diehl et al., 2016). Now it is already well known that TE are involved in epigenetic diversity, including changes in methylation patterns, histones’ modifications, birth of new microRNAs, and transgenerational epigenetic inheritance (Hosaka and Kakutani, 2018; Lanciano and Mirouze, 2018). It could be expected that the appearance of new niches formed by human and domesticated species promote TE activation and generation of new gene regulatory networks (Cho, 2018; Colino-Rabanal et al., 2018; Sherwin et al., 2019).
For example, in human brain tissue numerous TE-derived microRNAs (miRNAs) were found and many of those miRNAs were formed on the basis of LINE-2 (L2) retrotransposon, entering the human genome around 100–300 million years ago. L2-miRNAs formed on the basis of the 3′ end of the L2 consensus sequence shared very similar sequences, indicating that L2-miRNAs could target transcripts with L2s in their 3′ UTR. Accordingly, many protein-coding genes carry fragments of L2-derived sequences in their 3′ UTR serving as target sites for L2-miRNAs. As a rule, L2-miRNAs and their targets are ubiquitously expressed at low levels in multiple human tissues, suggesting a role for this network in buffering transcriptional levels of housekeeping genes (Petri et al., 2019).
It was also found that domestication process change miRNAs regulatory network. The comparison of miRNAs regulatory network between the now-extinct wild aurochs (Bos primigenius) and contemporary cattle (Bos Taurus) did show the changes in key network players (Braud et al., 2017). In contemporary cattle non-coding miRNAs became key regulators of spatiotemporal expression of gene targets that control mammalian growth and development. It is possible that in the domestication process selection of mutational changes in miRNAs and/or miRNA binding sites could have provided a mechanism to generate some of the traits that differentiate domesticated cattle from wild ancestors. The analysis of open reading frame sequences of 19,994 orthologous protein-coding gene pairs from extant Bos taurus genomes and extinct B. primigenius genome identified miRNA binding site polymorphisms in the 3′ UTRs of 1620 of these orthologous genes (Braud et al., 2017). These 1620 genes with altered miRNA binding sites represent candidate domestication genes and were mostly involved in pigmentation, fertility, neurobiology, metabolism, immunity, and production traits (including milk quality and feed efficiency). These results suggest that directional selection of miRNA regulatory variants was important in the domestication and subsequent artificial selection that gave rise to modern cattle (Braud et al., 2017).
Conclusion
Comparative analysis of domesticated species and the closely related wild animals led many researchers to agree upon an existence of “domestication syndrome” phenomenon. Generally, it is a group of systemic features that differentiate one species from others and are supposedly mostly related with suppressed neural crest development at the early stages of embryogenesis. As a rule all the studies of “domestication syndrome” were done with species pairs and limited sample sizes, consequently leading to unavoidable biases. Namely, in most cases with rare exceptions, for animal pairs under study, many different phenotypic characteristics were found to be associated with domestication syndrome. However, overall, we can dissect several repeated universal common properties that differentiate domesticated species from closely related wild animals. The most common features are elevated social activity and high phenotypic plasticity.
The most fascinating direction of contemporary domestication studies points toward strong connection between socializing behavior in mammalian species and brain-microbiome axe. Impressive microbiome differences were found between domesticated and closely related wild animals, in particular microbiome content in domesticated species is shifted toward higher abundance and diversity of Bifidobacterial species, helping with cellulose digestion and carbohydrate metabolism (Axelsson et al., 2013). These changes in microbiome content are apparently related with formation of common niche between domesticated animal and human and changes in the domesticated animal diet. The accumulated data suggest the interconnectedness of microbiome and virome, so the changes in one “ome” lead with necessity to changes in another. The remnants of viromes accumulated in genomes of mammalian species lead to significant changes in genomic structure. In particular, TE frequently modify gene regulatory networks by creating new miRNAs and their targets. Thus, the leading factor of universal features for domestication syndrome in different species could be changes in microbiome and, as a consequence, changes in regulatory networks of multicellular organism.