
Abstract
The tumor microenvironment (TME) plays a significant role in the occurrence, development, and prognosis of lymphomas. The mechanisms of each TME component in lymphoma and related therapeutic strategies are reviewed in this article. In the immune-related TME, regulatory T-cells (Tregs), tumor-associated macrophages (TAMs), and other immune cells mediate immune suppression and evasion. In the non-immune-related TME, cancer-associated fibroblasts (CAFs) and extracellular matrix (ECM) are involved in tumor progression, drug resistance, and immune evasion. In addition, aberrant glucose, amino acid, and lipid metabolism in tumor cells not only sustain their rapid proliferation and survival but also reshape the TME to favor immune escape, thereby worsening patient outcomes. Various treatment strategies targeting the components of the TME and metabolic abnormalities have brought new hope for the treatment of lymphoma. Further in-depth investigation into the intricate crosstalk between the TME and lymphoma cells will be instrumental in developing more effective, personalized precision therapies to improve patient survival.

Keywords
Introduction
The tumor microenvironment (TME) is the cellular environment in which tumors or cancer stem cells exist. It encompasses the non-cancerous cell components within the tumor and the molecules they produce and release. The acquisition and maintenance of cancer hallmarks, such as sustained proliferative signaling, resistance to cell death, induction of angiogenesis, activation of invasion and metastasis, pro-tumor inflammation, and immune evasion, depend, to varying degrees, on the TME 1 . TME can be divided into immune-related and non-immune-related TME. Immune-related TME mainly consists of immune cells and cytokines, including regulatory T-cells (Tregs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and others. These cells mediate the immune-inhibiting microenvironment and immune evasion. The non-immune-related TME is primarily composed of fibroblasts and other non-immune cells, including cancer-associated fibroblasts (CAFs), extracellular matrix (ECM) and various secreted factors, which provide structural support and pro-growth signals for the tumor. The complex interactions among these components collectively drive tumor progression2,3. However, one of the core engines driving these interactions is metabolic reprogramming. The intense competition for limited nutrient resources and metabolic adaptations between cancer cells and host cells within the TME fundamentally reshapes the local environment. This process not only meets the energy demands of rapid tumor growth but also suppresses effective antitumor immunity. Therefore, a deep understanding of the metabolic landscape of the TME is crucial for unraveling the essence of tumor biology and for developing novel therapeutic strategies.
Beyond its biological complexity, the TME holds profound therapeutic significance; targeting its unique metabolic vulnerabilities offers a promising avenue to disrupt tumor–stroma crosstalk, reverse immune suppression, and enhance the efficacy of existing treatments. This review aims to provide a high-level overview of the metabolic landscape within the TME. It will focus on elucidating how metabolic reprogramming acts as a central hub coordinating cellular interactions in the TME, thereby influencing tumor fate, and will explore its potential as a therapeutic target.
Methodology of study selection
Literature search and selection criteria
This review systematically retrieved relevant studies on the TME and metabolic reprogramming in lymphoma published from the establishment of databases to April 2025 from PubMed. The search keywords included: lymphoma, tumor microenvironment, metabolic reprogramming, glucose metabolism, lipid metabolism, amino acid metabolism, tumor-associated macrophages, myeloid-derived suppressor cells, cancer-associated fibroblasts, and other related terms. Meanwhile, the reference lists of relevant reviews and high-quality studies were manually searched to supplement important literature not included in the databases.
Inclusion criteria
Study type: Basic experimental studies, clinical studies, meta-analyses, systematic reviews, and other original English studies with complete data and clear conclusions.
Research objects: Studies focusing on the functional mechanisms of TME components, metabolic abnormalities, and targeted therapeutic strategies in lymphoma.
Research content: Studies involving the regulatory roles of immune cells and non-immune components in the TME; studies on the association between abnormal glucose, amino acid (AA), lipid, and other core metabolic pathways and lymphoma occurrence, progression, drug resistance, and immune escape; studies on therapeutic strategies targeting TME components or metabolic abnormalities.
Literature quality: For basic experimental studies, those with clear grouping, sufficient sample size, and repeatable results were included; for clinical studies, those with complete patient information, clear outcome indicators, and rigorous follow-up were included; for meta-analyses/systematic reviews, those with standardized search strategies and strict inclusion/exclusion criteria were included.
Exclusion criteria
Duplicate published literature, literature with incomplete data, or literature from which core information cannot be extracted. Studies whose research objects are other hematological malignancies or solid tumors not related to lymphoma, and without extended analysis on lymphoma. Literature with unclear research design, lack of positive/negative controls, or unreliable experimental results; case reports, conference abstracts, and thesis literature with insufficient depth. Reviews were only used to sort out references and cite core conclusions, not for direct data extraction and analysis.
Literature screening and data extraction
Two researchers independently completed literature retrieval, initial screening of titles/abstracts, full-text re-screening, and data extraction. Disagreements were resolved through consultation with a third researcher. The core extracted information included: lymphoma subtype, research model (cell line/animal model/clinical sample), key research findings, regulatory molecules/targets, intervention measures (drugs/genetic manipulation), and experimental/clinical outcomes. Finally, a total of 147 literatures were included in this review.
Tumor-associated macrophages
TAMs are abundantly present in the TME of most cancer types and are typically associated with poor clinical prognosis in cancer patients. The known pro-tumorigenic effects of TAMs include stimulating pro-tumor inflammation, promoting the immune escape of cancer cells, facilitating angiogenic transformation, and accelerating the invasion and metastasis of tumor cells1,4,5.
Polarization of TAMs
Key mechanisms driving TAM polarization
Macrophages exhibit two different polarization activation states, namely classically activated M1-type TAM (M1 TAMs) and alternatively activated M2-type TAM (M2 TAMs) 6 . M1 TAMs mainly exert pro-inflammatory, anti-bacterial, and antitumor functions by secreting pro-inflammatory cytokines, such as interleukin (IL)-1, IL-6, reactive oxygen species/reactive nitrogen species (ROS/RNS), and tumor necrosis factor (TNF)-α 7 . The TME often coerces them into adopting the M2 TAMs. This transition is frequently triggered by Th2-derived signals (IL-4, IL-13) and elevated CSF1R expression on macrophages, engaging critical signaling hubs, such as TLR4/NF-κB and IL-4/IL-13/STAT6 8 . Once polarized, M2 TAMs secrete reparative factors—including Th2 cytokines (IL-4, IL-10, IL-13), matrix metalloproteinases (MMPs), and tumor growth factor (TGF)-β—that fuel tumor growth and suppress immunity via diverse signaling networks 3 .
Evidence in cancer and lymphoma
Specific chemokine axes act as potent drivers of this pro-tumorigenic reprogramming. For instance, CCL2, abundantly secreted by tumor cells, recruits monocytes via the CCL2/CCR2 axis; upon binding, CCR2 activates intracellular cascades (PI3K/AKT, MAPK/p38, JAK/STAT3) that accelerate tumor initiation and progression 9 . Similarly, in B-cell lymphoma, genetic alterations in CREBBP or EP300 reduce H3K27 acetylation, thereby unleashing the NOTCH pathway to upregulate CCL2/CSF1, which further promotes proliferation and M2 polarization 10 . The CCL3-CCR1 axis also plays a pivotal role: high CCR1 expression on monocytes, combined with tumor-derived CCL3, not only facilitates cell migration but actively skews macrophage programming toward the immunosuppressive M2 phenotype while inhibiting M1 responses, thus remodeling the TME 11 .
Spatial heterogeneity and functional implications
The functional identity of TAMs (specifically CD163+ macrophages) is not uniform but varies significantly based on their spatial proximity to tumor cells. In tumor-rich areas (e.g. regions closely adjacent to tumor cells), CD163+ macrophages upregulate proteins related to apoptosis, DNA repair, and proliferation, including Bcl-2-interacting mediator of cell death (BIM), Bcl-2-associated agonist of cell death (BAD), and p53. In tumor-sparse areas (e.g. lymphoid tissues around the tumor), CD163+ macrophages exhibit higher levels of immunoregulatory proteins, such as VISTA and B7-H3, suggesting their active immunosuppressive function 12 .
TAMs promote tumor progression
TAMs produce various mediators that promote tumor progression, such as growth factors and cytokines, which support tumor cell proliferation, nuclear factor (NF)-κB-mediated anti-apoptotic factors, and angiogenic growth factors, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), TGF-β, and fibroblast growth factor 2 (FGF-2) 13 . They also promote tumor angiogenesis and vascular maturation by secreting semaphorin 4D (Sema4D), while REDD1-mediated mTOR inhibition may further influence tumor angiogenesis 6 . TAMs also produce other factors that regulate tissue structure and promote tumor cell migration, invasion, and metastasis, including MMP2, MMP9, cathepsins, CCL18, and CYP4A, which facilitate ECM degradation and tumor dissemination 8 .
In addition, TAMs express a variety of classical immune checkpoint inhibitors (ICIs), such as programmed death ligand 1 (PD-L1), and immunosuppressive enzymes, such as indoleamine 2,3-dioxygenase (IDO) and IL-4 induced gene 1 (IL-4I1). These molecules are involved in the catabolism of tryptophan (Trp) and phenylalanine, contributing to an immunosuppressive TME by inhibiting immune cell activity and indirectly suppressing T-cell function through the recruitment of Tregs and other immunosuppressive cells13,14. In natural killer/T-cell lymphoma (NKTCL), TAMs interact with tumor-infiltrating T-cells and exert immunosuppressive effects through CD80/CD86–CTLA4, PD-L1/PD-L2–PD-1, and LGALS9–HAVCR2 interactions, indicating their ability to inhibit T-cell immune function. Immunofluorescence staining revealed that PD-L1-expressing TAMs (CD206+ PD-L1+) were physically juxtaposed with CD8+ T-cells in NKTCL tumor tissues, further confirming the interaction between TAMs and T-cells and their immunosuppressive function
15
. TAMs can also inhibit cytotoxic CD8+ T-cell responses by metabolizing
TAMs in different types of lymphomas
Key mechanism: TAMs drive immunosuppression and tumor growth
In peripheral T-cell lymphoma (PTCL), high GATA3 expression signals a poor prognosis by fostering a Th2-like environment rich in cytokines such as IL-4, IL-5, IL-10, and IL-13 17 . These molecules act as architects of immune evasion, polarizing macrophages toward the pro-tumorigenic M2 phenotype 18 . Once established, M2 macrophages fuel angiogenesis via VEGF secretion while simultaneously engaging in an autocrine loop: they release IL-10 and TGF-to upregulate their own PD-L1 expression. This surface protein then binds to programmed cell death protein 1 (PD-1) on T-cells, effectively silencing their antitumor activity and creating a sanctuary for tumor cell survival (Figure 1).

Composition of the TME. TAMs inhibit the functions of tumor-infiltrating T-cells through their interactions with these T-cells. TAMs secrete various mediators that promote tumor progression. TAM, tumor-associated macrophage. (This figure was created by the authors using Microsoft PowerPoint 2019).
Evidence across lymphoma subtypes
A consistent narrative emerges across diverse lymphomas: abundant macrophage infiltration often heralds dismal outcomes.
T-cell lymphomas: In angioimmunoblastic T-cell lymphoma (AITL), a dominance of M2 macrophages closely tracks with poor prognosis. Similarly, in adult T-cell leukemia/lymphoma (ATLL), the presence of CD163+ TAMs serves as a stark indicator of unfavorable clinical trajectories 17 . Broadly, the density of CD163-positive macrophages inversely correlates with survival rates.
Follicular lymphoma (FL): Here, TAMs play a dual role. They can actively nurture FL B cells by crosslinking oligomannosylated B cell receptor (BCR) with Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), triggering sustained, albeit weak, signaling that bolsters tumor growth. In contrast, TAMs can phagocytose tumor B cells. However, FL B cells express CD47 in large quantities, which inhibit the phagocytic and inflammatory functions of macrophages expressing signal regulatory protein α (SIRPα), allowing tumor cells to evade macrophage-mediated clearance and facilitating immune escape process 14 .
In Hodgkin’s lymphoma (HL), an increased quantity of TAMs often indicates poor patient prognosis. The interaction between HL cells and TAMs involves the regulation of various signaling pathways, including the Jak/STAT signaling pathway, and the effects of cytokines, such as IL13, which further influence tumor progression 19 . In classical HL (CHL), the presence of TAMs is associated with patient survival. In a study involving patients with CHL, the TAM-enriched TME phenotype remained consistent from diagnosis to early relapse biopsy, with significant enrichment of CD163-positive macrophages in early relapse samples, suggesting an association with CHL recurrence and prognosis 20 .
Aggressive B-cell lymphomas: A meta-analysis demonstrated that a high density of M2 TAMs in the TME strongly predicts of poor prognosis in patients with diffuse large B-cell lymphoma (DLBCL) 21 . Similarly, in primary central nervous system lymphoma (PCNSL), an increased TAM levels are associated with shorter overall survival (OS) and progression-free survival (PFS). Increased TAMs levels may contribute to the poor prognosis of PCNSL22,23.
Therapeutic strategies targeting TAMs
Key mechanism: disrupting macrophage recruitment and survival
The CSF1R signaling pathway, activated by colony-stimulating factor 1 (CSF1), serves as a primary driver for TAM abundance and immunosuppressive activity. In mantle cell lymphoma (MCL), inhibiting this receptor eliminates CD163+ TAMs; specifically, the CSF1R inhibitor PLX3397 (pexidartinib) significantly reduces M2 TAMs viability. Furthermore, targeting the CCL2/CCR2 signaling axis offers a feasible immunological intervention, as CCR2 antagonists can reduce tumor growth and spread 24 . Similarly, in MCL, immunofluorescence analysis revealed that the CCR1 inhibitor treatment group demonstrated a significant reduction in M2 TAMs and an increase in CD8+ T-cell infiltration, indicating that inhibition of CCR1 could reduce the CCL3-CCR1 paracrine signaling axis between the tumor and macrophages, thereby alleviating the tumor burden 11 .
Therapeutic relevance: reprogramming phenotype and enhancing phagocytosis
What strategies exist to switch TAMs from a pro-tumor (M2) to an antitumor (M1) state or boost their engulfment capabilities? PI3Kγ is an important promoting factor for immunosuppression. Inhibiting PI3Kγ could promote TAM polarization toward the M1 phenotype, enhance immune response, and inhibit tumor growth and metastasis. PI3Kγ-specific inhibitors, such as IPI-549, are particularly applicable for tumors with high M2 TAM infiltration25,26. TGF-β, an important immunosuppressive and metastasis-promoting factor, drives M2 polarization and tumor progression 27 . Thus, inhibiting the TGF-β signaling pathway can help enhance immune cell activity. Similarly, the VEGF/VEGFR axis contributes to tumor angiogenesis and immune regulation by recruiting and polarizing TAMs and impairing T-cell function. Combination anti-VEGF therapies with ICIs can remodel the TME and enhance immunotherapy efficacy 28 . In preclinical models of DLBCL, anti-CD47 antibodies can reduce tumor burden and when combined with rituximab, demonstrate a synergistic effect in promoting macrophage-mediated phagocytosis of lymphoma cells 29 . In cutaneous T-cell lymphoma (CTCL), anti-PD-L1 and lenalidomide promote the repolarization of M2 TAMs to M1 phenotypes, as evidenced by the upregulation of M1 markers, such as CD80; the downregulation of CD163, PD-1, and PD-L1; changes in cytokine/chemokine expression; and inhibition of JAK/STAT and NF-κB signaling pathway. Their combination can inhibit M2 TAM migration by downregulating chemokines and their receptors, enhance phagocytic activity of M2 TAMs, and promote T-cell proliferation, thereby enhancing antitumor immunity 30 .
Epigenetic drugs: therapeutic landscape and immune regulatory mechanisms
Various epigenetic drugs, such as DNA methyltransferase inhibitors (DNMTi), histone deacetylase inhibitors (HDACi), BET inhibitors, and EZH2 inhibitors, have been approved by the U.S. Food and Drug Administration (FDA) or are in clinical trials for treating hematological malignancies and solid tumors 10 . Tucidinostat (chidamide), a novel subtype-selective HDACi targeting class I HDAC1, HDAC2, HDAC3, and class IIb HDAC10, has been approved for treating relapsed/refractory PTCL 31 .
The level of histone acetylation is determined by the dynamic equilibrium between histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs add acetyl groups to lysine residues of histones, whereas HDACs remove them 32 . Specifically, HDAC2 inhibitors can induce IL28A and IL28B expression, stimulate STAT1, and upregulate genes involved in double-stranded RNA pathogen recognition receptors and Major Histocompatibility Complex class (MHC) I class antigen presentation, potentially increasing immune cell infiltration in the TME and enhancing response to PD-1 blockade 33 . Furthermore, Nguyen et al. 34 reported that HDAC3 selectively binds to activating transcription factor 3 (ATF3) in a deacetylase activity-dependent manner, thereby inhibiting TLR signaling. Since ATF3 is a stress-induced transcription factor essential for regulating immune responses and maintaining normal host defense mechanisms, its inhibition via HDAC3 enhances NK cell cytotoxicity against lymphoma cells and simultaneously upregulates the expression of chemokines, such as CXCL12, among which CXCL12 and CXCL10 most effectively recruit NK cell35,36. EZH2 inhibitors can enhance the immunogenicity of lymphoma, promote T-cell function, and improve the therapeutic efficacy of T-cell immunotherapy on lymphoma, providing a new strategy for improving immunotherapy outcomes in patients with B-cell lymphoma 37 .
Preclinical data illustrate considerable potential in overcoming immunotherapy resistance in B-cell lymphoma 32 . For instance, the HDACi OKI-179 could inhibit the growth of G1XP lymphoma cells, induce cell cycle arrest (stopping cells at the G0/G1 phase and reducing the proportion of cells in S phase), and induce apoptosis (increasing the production of cleaved caspase-3). OKI-179 upregulated PD-L1 expression in both the G1XP and OCI-Ly7 lymphoma cells, with a dose-dependent effect observed specifically in G1XP lymphoma cells 38 . Meanwhile, per preclinical studies in mice, HDACi can enhance the efficacy of ICIs in treating various cancer types, including hepatocellular carcinoma and lymphoma38,39.
Ongoing clinical trials are evaluating the synergistic effects of combining epigenetic-targeting agents with ICIs. For instance, the combination of DNMTi with drugs, such as anti-PD-1 and anti-PD-L1 could enhance the antitumor immune response and improve immunotherapy efficacy 40 . While these findings highlight promising therapeutic relevance, further investigation is needed to fully define the optimal targets and address remaining uncertainties regarding long-term outcomes and patient selection. Table 1 shows the therapeutic strategies targeting TAMs in the TME.
Therapeutic strategies targeting TAMs in the TME.

Myeloid-derived suppressor cells
The key mechanisms driving the expansion and function of MDSCs
MDSCs are classified into two main types: polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs), and monocytic myeloid-derived suppressor cells (M-MDSCs)8,41. Under physiological conditions, immature myeloid cells (IMCs) can differentiate into mature monocytes, dendritic cells, and granulocytes. However, in pathological environments, the differentiation and maturation of IMCs are inhibited, leading to the expansion of MDSCs. Various factors in the TME, such as those secreted by tumor cells and extracellular signals, can promote the transformation of IMCs into MDSCs 24 . Factors related to tumors, such as chronic inflammation, hypoxia, nutritional deficiency, and growth factors, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and cytokines, including IL-6, TNF, and VEGF, jointly promote the generation of MDSCs42,43. Once formed, MDSCs are recruited to the TME through CXCR2, CCR2, CCR5, and other chemokine receptors. PMN-MDSCs are mainly recruited by CXC chemokines, whereas M-MDSCs migrate depending on the CCL2-CCR2 axis 42 . Factors related to the immunosuppressive ability of MDSCs include arginase 1 (ARG1), inducible nitric oxide synthase (iNOS), TGF-β, IL-10, cyclooxygenase-2 (COX-2) and IDO. Notably, ARG1 starves T-cells of arginine—an AA essential for their activation—thereby halting T-cell proliferation and function8,44.
Evidence linking MDSCs to cancer progression and lymphoma prognosis
As heterogeneous myeloid populations that swell within the TME, MDSCs act as architects of tumor immune evasion, angiogenesis, and metastasis, while also fostering resistance to cancer therapies 42 . In DLBCL, clinical data reveal a stark correlation: the higher the percentage of circulating M-MDSCs in the peripheral blood (PB), the later the stage of DLBCL, the higher the International Prognostic Index (IPI) score, and the worse the survival period 45 . Furthermore, survivors of DLBCL bear lasting scars of chronic inflammation, characterized by persistent inhibitory MDSCs, sustained T-cell activation, and eventual T-cell depletion—signatures that endure for years post-therapy 46 . The frequencies of MDSCs in both the bone marrow (BM) and PB of patients with DLBCL were elevated. Genes involved in the nitric oxide (NO) pathway, reactive oxygen process, cellular oxidative detoxification, heme signal transduction, and regulation of the autophagy pathway were significantly upregulated in BM-MDSCs 47 .
The therapeutic implications and remaining questions
Targeting MDSCs has become a key strategy for improving the therapeutic effects of cancer treatments 42 . The unique molecular landscape of BM-MDSCs offers specific avenues for intervention; disrupting their oxidative stress or autophagy pathways could potentially dismantle the immunosuppressive microenvironment. Such approaches hold promise for boosting the success of CAR-T cell therapies and bispecific antibodies 47 .
Mechanisms and targeting strategies of MDSC-mediated immunosuppression in lymphoma
In relapsed/refractory DLBCL (R/R DLBCL), MDSCs promote angiogenesis through VEGF, inhibit the maturation and function of NKs, and induce macrophage polarization to the M2 phenotype by secreting IL-10 and TGF-β to suppress the innate immune response 48 . MDSCs survive in the TME through metabolic reprogramming and are adverse prognostic indicators of hematological malignancies. The adenosine triphosphate (ATP) and oxaloacetate metabolism induced by the β2-AR-STAT3 pathway is a key regulatory factor for mitochondrial adaptation in MDSCs 49
In the in vivo EL4 lymphoma model, blocking β2-AR signaling, inhibiting mitochondrial electron transport chain, or reducing oxaloacetate production could enhance doxorubicin efficacy, promote MDSCs apoptosis, and reduce ROS (mROS) generation 49 . These findings highlight metabolism-driven pathways as promising therapeutic leverage points.
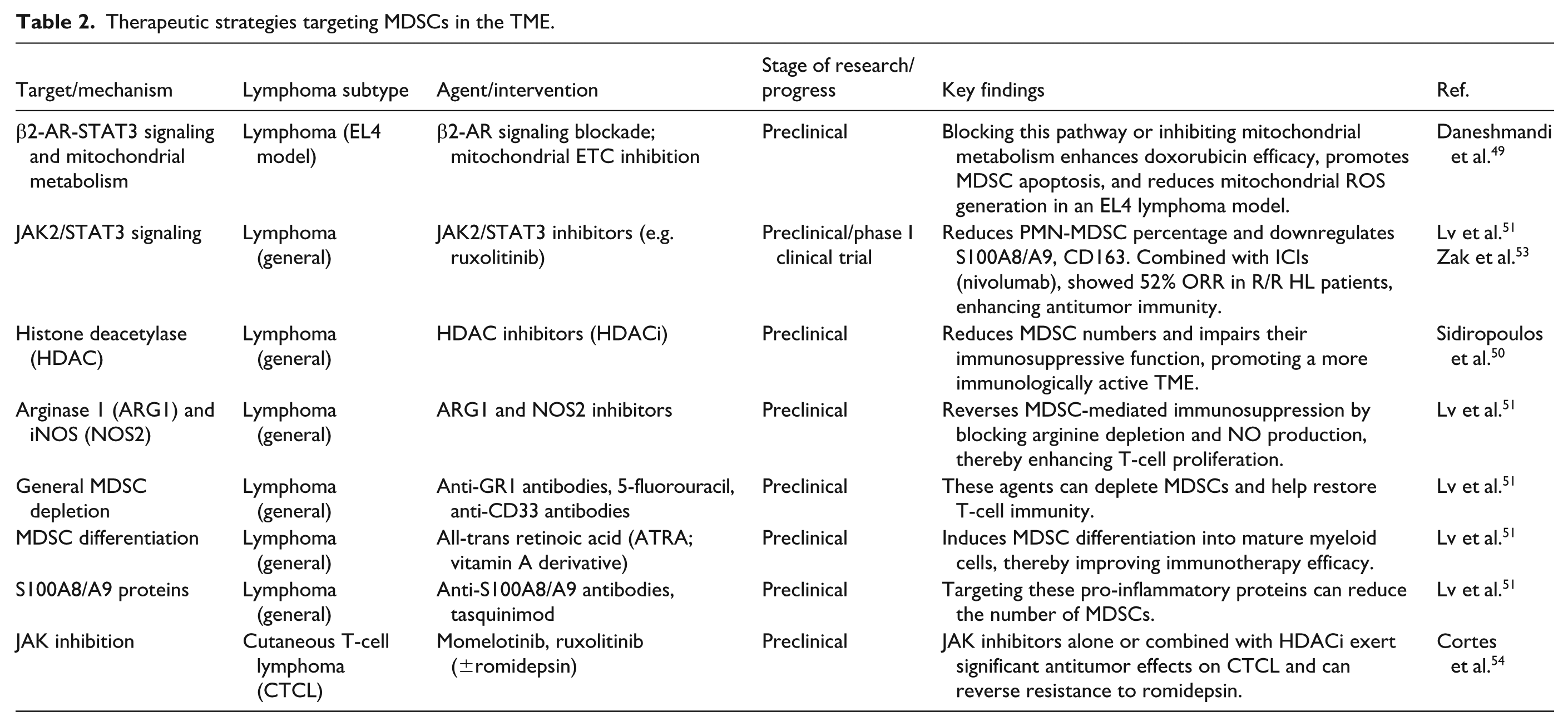
Multiple approaches effectively neutralize MDSCs. HDACi not only reduces the number of MDSCs but also impairs their immunosuppressive function, thereby promoting a more immunologically active antitumor TME 50 . The inhibition mechanism of MDSCs can be reversed using ARG1 and NOS2 inhibitors, phosphodiesterase-5 inhibitors, and bardoxolone methyl, thereby enhancing T-cell proliferation. Anti-GR1 antibodies, peptibodies, 17-DMAG, 5-fluorouracil, and anti-CD33 antibodies can be used to deplete MDSCs and restore T-cell immunity. Vitamins A, D3, and E induce MDSC differentiation. For instance, all-trans retinoic acid (ATRA) can improve immunotherapy efficacy. Antibodies against S100A8/S100A9, tasquinimod, zoledronic acid, and JAK2/STAT3 inhibitors can reduce the number of MDSCs51,52.
The JAK inhibitor ruxolitinib exemplifies translational potential. The JAK inhibitor ruxolitinib can significantly reduce the percentage of PMN-MDSCs in the blood. Ruxolitinib also downregulates the expression of MDSC-related markers such as S100A8, S100A9, and CD163, thereby weakening their suppressive function. In the presence of ICIs, ruxolitinib regulates myeloid cells, enhances antitumor immune responses, and preserves T-cell function. In a phase I clinical trial of patients with R/R HL, the objective response rate (ORR) of combination treatment with ruxolitinib and nivolumab was 52% (10/19), and some patients achieved a complete metabolic response or partial remission. The disease control rate was relatively high, indicating that JAK inhibitors can enhance the therapeutic effect of ICIs on lymphoma. Combination therapy showed good efficacy in patients with HL and provides a new direction for overcoming immune treatment resistance 53 . In CTCL, JAK inhibitors (e.g. momelotinib and ruxolitinib), alone or in combination with an HDACi (romidestat), exert significant antitumor effects on CTCL cell lines and can effectively reverse resistance in primary samples that are unresponsive to romidestat 54 . Table 2 shows the therapeutic strategies targeting MDSCs in the TME.
Therapeutic strategies targeting MDSCs in the TME.
CAFs and ECM
Key mechanisms: how CAFs drive tumor progression
Several different biomarkers are used to define CAFs, including but not limited to α-smooth muscle actin (α-SMA, also known as ACTC2), platelet-derived growth factor receptor α/β (PDGFRα/β), fibroblast-specific protein 1 (FSP-1, also known as S100A4), and fibroblast activation protein (FAP) 55 . CAFs play crucial roles in tumorigenesis, metastasis, angiogenesis, immune evasion, and drug resistance. For instance, they influence tumor progression by secreting regulatory molecules and extracellular vesicles (EAs) and remodeling the ECM 1 . Targeting the proline synthase PYCR1 (pyrroline-5-carboxylate reductase 1) in CAFs may inhibit the production of tumor-promoting extracellular matrix (TP-ECM) 56 .
Evidence in cancer and lymphoma: chemoresistance and immune evasion
Beyond shaping the physical tumor environment, CAFs actively confer chemoresistance to malignant cells. They stimulate cancer cell survival by delivering exosomes, promoting the epithelial–mesenchymal transition (EMT) in cancer cells, and eliminating chemotherapeutic drugs. CAFs are also involved in immune evasion and resistance to immunotherapy 1 . CAFs contribute to immune evasion by forming an immune barrier through the secretion of chemokines and TGF-β, which recruit and polarize TAMs. In addition, neovascularization in lymphoma acts as a functional barrier, facilitating immune tolerance 57 . In FL, CAFs play a significant role in the TME. They express various chemokines, such as CXCL12, CCL19, CCL21, and CCL2, which affect the growth of tumor B cells, recruit immune cells, and regulate TAM functions 14 .
Specifically, CXCL12 is a key factor that regulates the migration of lymphoma cells from blood to lymphoid tissues. It binds to its receptor, CXCR4, and activates downstream signaling pathways, promoting cell survival, metastasis, and chemotherapy resistance. CXCL12 also interacts with 2-arachidonoyl glycerol (2-AG) to regulate lymphoma cell chemotaxis. In chronic lymphocytic leukemia (CLL), 2-AG and CXCL12 jointly increase cell migration. In MCL cells, chemotaxis was reduced after combined treatment in most samples 58 . The efficacy of immunotherapy relies on the accumulation of antigen-specific CD8+ T-cells within tumors, a process governed by the CXCL12-CXCR4 axis via tumor-associated lymphatic vessels. Consequently, inhibiting CXCL12/CXCR4 signaling can boost the population of functional CD8+ T-cells inside tumors, improving tumor control 59 .
Therapeutic relevance: targeting exosomes and signaling axes
Exosomes (small extracellular vesicles, sEVs) are potential biomarkers for monitoring and exploring tumor development mechanisms. Exosomes enter the TME by carrying the CD98hc protein, promoting tumor cell proliferation and invasion and chemotherapy resistance (i.e. resistance to panitumumab), leading to disease progression. Inhibiting exosome generation can block this process and enhance the efficacy of chemotherapy, providing a new strategy for overcoming drug resistance 60 . Some studies highlighted that exosomes derived from DLBCL can promote tumor progression by transferring GP130 to macrophages, activating STAT3 signaling, and polarizing macrophages toward a tumor-promoting M2-like phenotype. Using exosomes in the TME and macrophages as therapeutic targets provides a new direction for precise clinical treatment of DLBCL 40 .
Exosomes modulate antitumor immunity and serve as therapeutic vehicles
Tumor-derived exosomes (TDEs) secreted by lymphoma cells carry PD-L1 and bind to PD-1 on the surface of T-cells, inducing T-cell apoptosis or functional exhaustion, and weakening the antitumor effect of CAR-T cells. In DLBCL, the level of PD-L1 in TDEs positively correlates with disease progression, and blocking the PD-L1 pathway reverses immunosuppression. Conversely, chimeric antigen receptor (CAR) exosomes are derived from CAR-T cells, carrying the same CAR as the parent cell and containing cytotoxic molecules, such as granzyme B and perforin, which can directly kill lymphoma cells expressing the corresponding antigen 61 . Leveraging the tumor-homing behavior of cancer cells, researchers have prepared activated macrophage-tumor chimeric exosomes (aMT-exos) that can accumulate in lymph nodes and tumors, activate immune responses, improve the TME, and effectively inhibit primary tumors, tumor metastasis, and postoperative tumor recurrence. This provides a new strategy for personalized immunotherapy 62 .
The role of the ECM in tumor progression and immune evasion
The ECM can be divided into the basement membrane and the interstitial matrix 63 . ECM serves as a supporting framework for tumor cells, creating a specific survival environment 64 . Abnormal degradation and deposition are associated with tumor progression and can affect the function of immune cells 63 . In HL, tumor and stromal cells can modify ECM properties 65 . The remodeling of ECM alters its physical properties and composition, creating favorable conditions for tumor cell migration. Circulating tumor cells (CTCs) upregulate the expression of certain ECM proteins, which support their survival, enable evasion of immune clearance, and facilitate formation of metastatic foci 64 (Figure 1). Table 3 shows the therapeutic strategies targeting CAFs and ECM in the TME.
Therapeutic strategies targeting CAFs and ECM in the TME.

Genetic alterations shaping the TME
Genetic characteristics play a decisive role in shaping the TME of lymphoma, particularly genetic abnormalities associated with antigen presentation. These abnormalities act like hidden barriers, aiding tumor cells in evading immune surveillance. Specifically, mutations or deletions in the β2-microglobulin (B2M) gene impair the assembly of MHC class I molecules, thereby preventing effective presentation of tumor antigens and allowing cancer cells to evade recognition by CD8+ T-cells. Meanwhile, mutations in the CD58 gene disrupt its critical interaction with CD2, inhibiting T-cell activation signaling. These two types of genetic events collectively reveal the frequent immune escape mechanisms in DLBCL and contribute to the formation of an ‘immune-desert’ cold TME 66 . Furthermore, abnormalities in epigenetic regulators, such as EZH2 mutations or CREBBP/EP300 deletions, suppress the expression of MHC molecules and antigen-processing-related genes (e.g. TAP1, LMP2) through histone modifications 67 . These early genetic alterations are closely associated with the impaired antigen-presenting function observed in FL precursor cells67,68.
At a broader level, driver genetic alterations (such as MYC proto-oncogene (MYC) amplification and TP53 mutation) not only regulate the secretion of chemokines to suppress dendritic cell maturation and T-cell infiltration but also promote the recruitment of M2-type TAMs and MDSCs, thereby reinforcing the immunosuppressive barrier69,70. Meanwhile, chromosomal deletions or translocations (e.g. 9p24.1 amplification) can lead to abnormal amplification of immune checkpoint molecules (e.g. PD-L1) or sustained activation of immune-activating pathways (e.g. NF-κB), further reshaping the immunosuppressive microenvironment 71 .
Targeted therapies directed against these genetic markers hold great clinical potential. For instance, EZH2 inhibitors can reverse MHC expression defects and restore antigen-presenting function, thereby enhancing the tumor-recognition capacity of ICIs or CAR-T cells—a strategy that has been established as an important therapeutic target for improving immune recognition 72 . Therefore, for lymphoma patients with B2M mutations, exploring combination therapies of “epigenetic modulators + tumor vaccine + PD-1 inhibitors” holds promise in compensating for the defect in antigen presentation and transforming the cold TME into a hot, immune-active battlefield.
Glucose metabolism
In normal cells, glucose uptake initiates a streamlined process where extracellular sugar is internalized and converted into pyruvate via glycolytic enzymes. Under aerobic conditions, this pyruvate enters the mitochondria to fuel the tricarboxylic acid (TCA) cycle, ultimately yielding carbon dioxide, water, and approximately 36 ATP molecules per glucose unit through oxidative phosphorylation (OXPHOS) 73 . Only in hypoxic environments, the cells rely on anaerobic glycolysis to convert pyruvate into lactate 74 .
Malignant cells, driven by the need for rapid proliferation, fundamentally alter their metabolic landscape compared to normal counterparts. Tumor cells have specific metabolic processes, such as the Warburg effect (aerobic glycolysis), which reprograms glucose metabolism, even under aerobic conditions. They utilize glycolysis, rather than mitochondrial OXPHOS, to obtain energy and metabolic precursors for biomass production75,76. This reliance on glycolysis is supplemented by critical branching pathways: the pentose phosphate pathway (PPP), glycogen synthesis from 6-phosphoglucose, the hexosamine biosynthesis pathway (HBP) derived from 6-phosphofructose, and the serine biosynthetic pathway (SBP) originating from 3-phosphoglyceric acid, all of which support tumor adaptation and expansion 77 .
Beyond energy production, glycolytic activity can reduce chemokine expression, limit immune cell infiltration, and upregulate PD-L1 by activating the NF-κB pathway, thereby inhibiting the immune response 78 . Specific evidence in lymphoma highlights that Latent Membrane Protein 1 (LMP1) activates the non-classical NF-κB pathway by competitively binding to Tumor Necrosis Factor Receptor-Associated Factor 3 (TRAF3), thereby promoting aerobic glycolysis in NKTCL and enhancing tumor cell proliferation, anti-apoptosis, and chemotherapeutic resistance 79 .
Aerobic glycolysis
Key mechanism: lactic acid reshapes the immune landscape
Tumor cells undergo aerobic glycolysis to produce lactic acid, which is subsequently transported out of the cells to the extracellular environment through the activation of monocarboxylate transporters on the cell membrane (especially MCT4), leading to acidification of the TME 80 . Lactic acid activates the mTOR pathway, thereby phosphorylating the transcription factor TFEB and inhibiting its nuclear translocation, which, in turn, suppresses the expression of vacuolar ATPase subunit (ATP6Vod2) in TAMs. Inhibition of ATP6Vod2 mediates lysosomal degradation of hypoxia-inducible factor 2-α (HIF-2α) and program TAMs in the TME as immunocytes promoting tumor growth 80 (Figure 2) Gpr132 (a G protein-coupled receptor) recognizes lactic acid on TAMs and also promotes the polarization of TAMs toward the M2 phenotype. Moreover, the expression of Gpr132 is inhibited by the peroxisome proliferator-activated receptor (PPARγ) transcription factor 73 . Beyond myeloid reprogramming, lactic acid also inhibits the functions of T-cells and NKs, while promoting the proliferation and activity of immunosuppressive cells, such as Tregs and MDSCs, thereby facilitating tumor immune evasion81,82.

Lactic acid produced by aerobic glycolysis of tumor cells is transported out of the cells by MCT4, thereby promoting TAMs polarization to M2 phenotype. (This figure was created by the authors using Microsoft PowerPoint 2019).
Evidence in lymphoma and metabolic competition
Clinical observations confirm that serum lactic acid levels increase progressively in patients with lymphoma 83 . The high glucose uptake and metabolism of tumor cells may lead to an insufficient glucose supply for immune cells, affecting their functions and weakening the body’s antitumor immune response81,82. In a high glucose environment, lactic acid could inhibit the function of conventional T-cells, but Tregs are resistant to the equivalent concentration of lactic acid in tumors. Their inhibitory functions and proliferation remain unaffected. Treatment with the PEPCK inhibitor 3MP revealed that lactic acid uptake and oxidation, along with upstream gluconeogenic reactions involving PEP, play important roles in the proliferation and inhibition of Tregs 84 . In DLBCL, the high-grade malignant B-cell subpopulation exhibits enhanced glycolytic activity. In DLBCL tissues with high glycolytic metabolism, the abundance of interferon (IFN)-TAMs (CD68+CXCL10+PD-L1+) increases, whereas the infiltration of CD8+ T-cells decreases, indicating an immunosuppressive microenvironment 85 .
Key enzymes in glycolysis
Various key enzymes involved in glycolysis, such as hexokinase 2 (HK2), pyruvate kinase (PK), and phosphofructokinase (PFK), are abnormally expressed in tumor cells86,87. Overexpression of HIF1-α is closely related to hematological malignancies, such as leukemia, lymphoma, and multiple myeloma 88 . In HL, HIF-1α is mainly expressed in the hypoxic side population (SP) of HL cells. HIF-1α triggers the expression of heme oxygenase-1 (HO-1), which scavenges mROS within SP cells and inhibits tumor cell differentiation, resulting in poor patient prognosis 89 . HIF-1α also promotes the secretion of VEGF by lactic acid and induces the polarization of TAMs toward the M2 type 73 . Overexpression of HIF-1α promotes the activity and migration of lymphoma cells. HIF-1α activates the transcription of CXCR4 by binding to the functional site HRE1 on the CXCR4 promoter, thereby activating the AKT/mTOR signaling pathway and promoting the proliferation and migration of lymphoma cells. Therefore, HIF-1α may serve as a potential therapeutic target for lymphoma 90 .
Hexokinase
Key mechanisms: the metabolic engine and feedback loops
HK has four isoenzymes, namely HK1, HK2, HK3, and HK4. HK1 is typically widely expressed, whereas HK2 is expressed only in adipocytes, bones, and cardiac muscles 91 . The expression of HK2 is increased in tumors, and its enhanced activity can promote glucose uptake and phosphorylation, providing more energy and synthetic raw materials for tumor cells, thereby promoting tumor cell proliferation81,92. HIF-1α can upregulate HK2 expression, which promotes glucose conversion into glucose-6-phosphate (G-6-P), thereby enhancing glycolytic flux. Meanwhile, the accumulation of a large amount of glycolytic intermediates can increase the level and activity of HIF-1α, forming a positive feedback loop, which further promotes aerobic glycolysis and maintains the survival and proliferation of tumor cells 93 . Beyond metabolism, HK2 exhibits non-classical functions, such as binding to mitochondrial pore proteins to inhibit apoptosis and directly maintaining tumor cell stemness94,95.
Evidence in lymphoma: dysregulation and prognostic value
In the context of lymphoma, HK2 serves not only as an important marker related to metabolic function but also holds potential prognostic value81,92. In PCNSL, the activity of HK2 increases, resulting in higher glycolytic activity of this tumor compared to normal lymphoid tissues 93 . Specifically, patients with PCNSL exhibiting normal immune function and positive Epstein–Barr virus (EBV) status activate the RelA/p65-HK2 signaling axis through different mechanisms, driving glycolysis and promoting tumor progression 96 . Similarly, in DLBCL, HK2 showed significantly high expression. Knockdown of HK2 can reduce tumor growth 97 . Conversely, HK3 shows a different pattern; mainly found in hematopoietic cells, it is upregulated during terminal differentiation in certain acute myeloid leukemia (AML) models. However, unlike HK2, HK3 deletion in AML cell lines does not alter basal glycolytic activity, suggesting a divergent role in metabolic regulation 98 .
Therapeutic relevance: targeting the axis
The dependency of tumors on HK2 offers promising therapeutic avenues. Knockdown of HK2 effectively curtails tumor growth in DLBCL 97 . Per another study, high HK2 expression is closely related to intracerebral tumor progression of PCNSL. Knockdown of RelA/p65 or inhibition of the NF-κB pathway (such as BAY11-7082) significantly reduced HK2 levels and inhibited tumor cell growth 96 . In AML, while HK2 knockout diminishes glycolytic capacity, the deletion of HK3 significantly enhanced the sensitivity of AML cell lines to cell death induced by ATRA. During the ATRA-induced differentiation process, HK3-deficient (HK3-null) AML cells exhibited an accumulation of ROS and DNA damage 98 . Studies revealed that a novel isomerase isoform of hexokinase domain containing 1 (HKDC1) is involved in glucose homeostasis and tumor development. HKDC1 exhibits high sequence similarity with the other four HK isoforms, with only the last eight C-terminal AAs showing differences. In SNK6 cells, overexpression of HKDC1 promotes cell proliferation, while knockdown of HKDC1 inhibits cell proliferation and increases cell apoptosis, indicating that HKDC1 plays a dominant role in the growth of extranodal natural killer/T-cell lymphoma (ENKTL) 99 . Critically, ablating HKDC1 can inhibit tumor sphere formation (clusters of cancer cells), cell migration, and cancer cell invasion, as well as reduce the expression of stem cell markers, such as ABCG2, Oct4, Sox2, CD13, and CD133. Re-expression of HKDC1 rescued these phenotypes, indicating that its crucial role in promoting cancer cell proliferation, stem cell maintenance, and tumor growth 100 .
Pyruvate kinase
Studies have identified four tissue-specific isoforms of PK expressed in mammals: PKL, PKR, PKM1 and PKM2. PKR is mainly expressed in red blood cells. PKL is expressed only in the liver, kidneys, and intestines. PKM1 is expressed in terminally differentiated tissue types, such as the brain and muscles and PKM2 is expressed in proliferative tissues and tumor cells 101 . Low PKM2 activity can alter glycolytic flux, facilitating the entry of upstream intermediates into the biosynthetic pathway, thereby supporting tumor growth and proliferation 81 . At the molecular structure level, PKM2 can aggregate into tetramers and dimers102,103. The dimer PKM2 can reportedly promote the proliferation, migration, and cell-cell ECM adhesion of endothelial cells, thereby promoting tumor growth. The signaling pathway mediated by insulin-like growth factor (IGF)-1/IGF-IR enables PKM2 to translocate into the cell nucleus. The PKM2 in the nucleus (nPKM2) activates HIF-1α and NF-κB, upregulates the expression of VEGF, and promotes tumor angiogenesis 101 . Furthermore, nPKM2 promotes EMT by interacting with TGIF2, an inducer of TGF-β, and downregulating the expression of E-cadherin. This protein plays a role in the malignant invasion and metastasis of cancer cells. The extracellular PKM2 (sPKM2) promotes cancer cell migration through various signaling pathways, such as PI3K/Akt and Wnt/β-catenin 104 .
Notably, MYD3/H3K4me3/PKM2 axis plays a significant role in DLBCL. Lysine histone methyltransferase (SMYD3) promotes the transcription of PKM2 through H3K4me3, thereby facilitating the proliferation and aerobic glycolysis of DLBCL cells. SMYD3 is significantly overexpressed in DLBCL tissues and is associated with poor prognosis and chemotherapy resistance, making it a potential prognostic biomarker and therapeutic target for DLBCL 105 .
Phosphofructokinase-1
Key mechanism: PFKP drives tumor aggression
PFK1, a key rate-limiting enzyme in the glycolytic process, exists in three tissue-specific isoenzymes: muscle type (PFKM), liver type (PFKL), and platelet type (PFKP). PFKP is highly expressed in various malignant tumors, including breast cancer, hepatocellular carcinoma, prostate cancer, glioblastoma, and leukemia. Its high expression positively correlates with tumor progression 106 . PFKP Y64 phosphorylation promotes glycolysis in EGFR-activated enhanced tumor cells in a β-catenin transcriptional activation-dependent manner. It also enhances β-catenin S552 phosphorylation mediated by AKT, thereby promoting its nuclear translocation and downstream gene expression and ultimately promoting the proliferation, migration and invasion of tumor cells 107 .
Evidence in cancer and lymphoma: metastasis and metabolic reprogramming
Beyond local growth, PFKP plays an important role in tumor metastasis. CXCR4 is crucial for the migration of normal hematopoietic stem cells, progenitor cells, and lymphocytes and is implicated in the metastasis of leukemia and cancer cells. Nuclear PFKP promotes CXCR4 expression, thereby enhancing the migration of T-cell acute lymphoblastic leukemia (T-ALL) cells 108 . Moreover, in T-cell lymphoma, deletion of the Pdcd1 gene encoding PD-1 leads to the upregulation of PFK-1 and other key glycolytic enzymes, including HK-2, aldolase A, and enolase 1. This enhances glucose uptake and metabolism in tumor cells, thereby increasing energy carrier production, supporting rapid proliferation and malignant transformation, and ultimately promoting tumor progression 109 . In addition to the key enzymes involved in glycolysis, the expression of glycogen synthase kinase 3 (GSK-3) is increased in DLBCL cell lines and is associated with enhanced cell survival and proliferation. Inhibiting GSK3 can reduce the proliferation of lymphoma cells 97 .
Therapeutic relevance: targeting glycolytic branch pathways
In addition to glucose oxidation through glycolysis, which generates acetyl-CoA and lactic acid, glycolytic branch pathways also play a role in tumor growth and adaptation. These branch pathways transfer glucose carbon flux from glycolytic processes at various levels to several anabolic pathways, including the PPP, glycogen synthesis, HBP, and SBP. The metabolites produced by these pathways can be utilized in various metabolic processes, enabling tumor metabolic reprogramming with special flexibility 77 . PPP plays a significant role in lymphoma cells. CLL cells divert large amounts of glucose flux into the PPP. Moreover, PPP protects B-ALL cells from oxidative stress. Transcription factors, such as PAX5 and IKZF1 negatively regulate the expression of the key PPP enzyme G6PD, thereby influencing the metabolic state of tumor cells. Meanwhile, SBP has been identified as a key metabolic node regulating the proliferation of malignant B cells 110 . Its activity is upregulated during B-cell activation and germinal center reaction. In human germinal center B-cell lymphomas, the key enzymes Phosphoglycerate dehydrogenase (PHGDH) and PSAT1 in SBP are overexpressed. Importantly, pharmacological inhibition of PHGDH effectively suppresses lymphoma cell proliferation, highlighting a promising therapeutic avenue 77 .
Key mechanisms: transcriptional regulators and transporters drive tumor metabolism
Members of the PRDM family (including PRDI-BF1 and RIZ homologous domains) are sequence-specific transcriptional regulators that are often dysregulated in cancer. PRDM15 regulates the transcription of key genes in lymphoma cells and is involved in two metabolic pathways that are crucial for the survival of tumor cells: the PI3K/AKT/mTOR signaling pathway (InsR and Igf1R) and glycolysis (Pkm, Pfkp, and Eno3). Deletion of PRDM15 leads to downregulation of the transcription of these genes, thereby affecting the activity of metabolic pathways 111 . In terms of metabolic regulation, oncogenic HSP90 simultaneously supports glycolysis and mitochondrial respiration. In lymphoma cells, oncogenic HSP90 more effectively utilizes the metabolites required for synthesizing macromolecules, which is conducive to maintaining cellular biosynthetic activity. The inhibition of oncogenic HSP90 reduces glucose uptake and lactate secretion in DLBCL cells, impairs glycolytic function, and increases glycolytic reserves. It also reduces glutamine (Gln)-induced mitochondrial oxygen consumption, thereby lowering basal oxygen consumption rate and respiratory parameters 112 .
Evidence in cancer: the role of GLUT proteins in tumor progression
Glucose transporter (GLUT) is responsible for transporting glucose into the cells. Among them, GLUT1, GLUT3, and GLUT4 are overexpressed in various tumors. Oncogenes such as c-MYC, KRAS, and YAP can upregulate the expression GLUT1, promoting glucose uptake by tumor cells. P53, a tumor suppressor protein, inhibits the transcription of GLUT1, GLUT3, and GLUT4. P53 inactivation leads to an increase in GLUT3 expression, thereby increasing the rate of tumor glycolysis (Figure 2) 92 . GLUT1 is widely expressed in various tumor cells, and its overexpression increases glucose uptake and promotes tumor cell growth and proliferation 113 . Single-cell RNA sequencing (scRNA-seq) and other techniques have revealed that GLUT1 is highly expressed specifically in M2 TAMs, indicating their strong capacity for glucose uptake. In M2 TAMs, an increase in glucose utilization increases the level of OGT-catalyzed cleavage of tissue proteinase B by lysosomes (cathespin B O-GlcNAcylation), thereby increasing the mature form of tissue proteinase B and promoting cancer metastasis and chemotherapy resistance 114 . Studies have shown that CAR-T cells overexpressing GLUT1 exhibit stronger metabolic adaptability in vivo, characterized by the simultaneous upregulation of genes related to glycolysis and mitochondrial respiration and an increased ability to utilize glucose. Moreover, GLUT1 overexpression helps maintain the memory phenotype of CAR-T cells 115 .
Therapeutic relevance and targets: exploit metabolic vulnerabilities
Based on the high glucose uptake characteristics of tumor cells, positron emission tomography (PET) imaging with 18F-labeled fluorodeoxyglucose (FDG) can be used to detect glucose uptake by tumor tissues. This method can be used for tumor diagnosis and monitoring therapeutic responses 116 . Various therapeutic strategies and drugs targeting glucose metabolism have been developed based on abnormal glucose metabolism in tumor cells. For instance, inhibitors of GLUTs (e.g. STF-31 and BAY-876) and key enzymes involved in glycolysis (e.g. 3-bromopyruvate, HK2, and LDHA inhibitors) have been studied and applied in tumor treatment 81 . Table 4 shows the therapeutic strategies targeting glucose metabolism in the TME.
Therapeutic strategies targeting glucose metabolism in the TME.

AA metabolism
Cellular uptake of AA requires the participation of AA transporters (AATs). AA plays significant roles in key pathways, such as cell growth, metabolism, and immunity, including the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway, gluconeogenesis, and proliferation and differentiation of immune cells 117 .
AA metabolism in tumor cells is characterized by a decreased breakdown of AA and enhanced protein synthesis. This metabolic reprogramming leads to an increased demand of the tumor cells for AA, such as arginine, Gln, and Trp, thereby altering their levels in the TME 73 .
In T-ALL cells, after treatment with fibroblast growth factor receptor 1 (FGFR1) inhibitors, many metabolic pathways related to survival, especially AA metabolism and its metabolites, are upregulated. Furthermore, inhibiting key metabolic genes (e.g. ASNS, ASS1, SLC1A5, and PHGDH) directly suppresses the activity of mTORC1 and enhances the sensitivity of T-ALL cells to FGFR1 inhibitors. These findings suggest that targeting AA metabolic pathways may be a potential strategy for overcoming drug resistance in tumors 118 .
Arginine
Key mechanism: tumors hijack arginine metabolism
Arginine is required for tumor growth. Arginine synthesized intracellularly is largely derived from the urea cycle. Ornithine and citrulline are converted into arginine by the catalytic activities of ornithine transcarbamylase (OTC), argininosuccinate synthetase-1 (ASS-1), and argininosuccinate lyase. Notably, tumor cells significantly increase their consumption of arginine 73 .
Evidence in cancer and lymphoma
In various tumors, the transcription of ASS-1 is suppressed, resulting in the dependence of tumor cells on exogenous arginine 117 . Notably, PTCL exhibits arginine auxotrophy and relies on the arginine transporter SLC3A2 for arginine uptake from the environment to sustain proliferation and escape immune surveillance 119 . Similarly, in lymphoma cells, increased arginase activity results in a significant decrease in plasma arginine levels 120 . Among them, elevated level of arginase-1 leads to the decomposition of L-arginine into urea and L-ornithine, as reported in many cancers 73 . This makes lymphoma cells highly dependent on extracellular arginine and arginine transporters to maintain biological functions, such as proliferation, survival, and protein synthesis 120 . Furthermore, in PTCL, arginine promotes tumor progression by promoting the synthesis of new RNA and enhancing OXPHOS 119 . In lymphoma, the arginine deprivation-mediated immunosuppressive TME mainly functions through two enzymes: nitric oxide synthase (NOS) and arginase. Both are conducive to the immune escape of cancer cells. Hematopoietic system malignant cells, MDSCs, and TAMs express NOS2 and/or arginase, deprive the TME of arginine, and inhibit T-cell proliferation and cytotoxicity 120 . Moreover, arginine metabolism helps maintain the immunosuppressive function of MDSCs 121 .
Therapeutic relevance and targets
Arginine deprivation therapy could be used for treating related tumors highlighting the potential therapeutic value of arginase inhibitors. Moreover, protein arginine methyltransferases (PRMTs) are associated with tumor stem cell characteristics, and their inhibition suppresses tumor cell proliferation and induces apoptosis 117 . Promisingly, Quinacrine, when used in combination with HDACi, showed synergistic antitumor effects both in vitro and in vivo, providing a new combined strategy for the treatment of PTCL 119 .
Glutamine
Key mechanisms of Gln metabolism
Gln is one of the most abundant AA and participates in various cellular metabolic processes. After entering the cells through AATs, such as ASCT2, it is metabolized into glutamate and ammonium by enzymes, such as glutaminase (GLS) and ultimately converted into lactic acid and ATP93,117. Gln can also be converted into glutathione to maintain the redox balance within the cells 73 . Tumor cells have a high demand for Gln, and their metabolism is regulated by various oncogenes and tumor suppressor genes, such as c-MYC and P53 122 . MYC could induce the transcription of AATs, such as Asct2 (SLC1A5) and LAT1 (SLC7A5), thereby upregulating the expression of AATs in MYC-expressing B cells and increasing AA uptake 123 . MYC can also upregulate the expression of GLS1, while P53 promotes mitochondrial respiration and TCA activity by upregulating the expression of GLS-2, thereby regulating the redox balance and proliferation of cells 93 . Furthermore, SLC7A5 exchanges Gln and leucine to activate the MTORC1 pathway, which is a crucial regulatory factor for cell growth in hematological malignancies 124 .
Evidence in lymphoma and the TME
In the TME, Gln metabolism affects the functions of immune cells, and tumor cells compete with immune cells for Gln uptake. Inhibiting Gln metabolism promotes the differentiation of CD4+Th1 cells and CD8+ T-cells, and inhibits Th17 cell differentiation. Gln metabolism-related inhibitors can regulate T-cell function and enhance antitumor immunity 125 . In Burkitt’s lymphoma (BL), high expression of SLC1A5 and SLC7A5 promotes disease progression, with their expression being dependent on Gln availability. Their upregulation is triggered by the abnormal activation of MYC in BL, thereby promoting cell dependence on Gln for growth and survival 120 . In MYC-induced BL cell lines, Gln is metabolized through TCA under hypoxic conditions. Inhibition of this process can effectively suppress tumor progression. In DLBCL, the transcription factor NRF2 (NFE2L2) correlates with advanced clinical stages and IPI scores, driving metabolic reprogramming that heightens reliance on the Gln pathway and susceptibility to GLS inhibition 126 . Similarly, in MCL, resistance to ibrutinib is linked to Gln dependence and the necessity of the transporter ASCT2, where elevated GLS expression fuels both proliferation and drug resistance 127 . In CLL, Gln uptake underpins resistance to venetoclax (VEN) induced by CD40/BCR stimulation, highlighting AA metabolism as a central pillar of resistance mechanisms across these malignancies 128 .
Therapeutic relevance and emerging targets
Targeting the Gln metabolic axis offers a promising strategy to overcome therapeutic resistance and inhibit lymphoma growth. The GLS inhibitors, BPTES and CB-839, can inhibit the proliferation of lymphoma cells, and CB-839 can impair glutathione production 93 . In MCL, the highly specific GLS inhibitor, taregenastat (CB-839), inhibits the growth of MCL cells and simultaneously enhances the efficacy of ibrutinib and venetoclax(VEN) in vitro and in vivo 127 . Likewise, in CLL, the Gln transporter inhibitor V9302 mitigates VEN resistance by blocking Gln uptake, effectively re-sensitizing cells to treatment 128 .
Tryptophan
The central role of Trp in biology
Trp is a fundamental component of protein synthesis and serves as a precursor for many important biological molecules. Its metabolic pathways include the serotonin and kynurenine (Kyn) pathways. These metabolites play significant roles in immune regulation, neurotransmission, and cell growth 129 .
Tumors exploit Trp metabolism to evade immunity
Trp can be rapidly internalized by tumor cells through the cell membrane. Most solid tumors express high levels of indoleamine 2,3-dioxygenase (IDO1) and tryptophan 2,3-dioxygenase (TDO2), which are the two main dioxygenases involved in the decomposition of Trp into Kyn. Trp-deficiency syndrome (TDS) is caused by the metabolism and utilization of Trp by tumor cells in the TME 73 . In lymphoma, IDO1 + tumor cells catalyze the degradation of Trp through IDO1, resulting in the reduction of Trp in the TME and the accumulation of metabolites, such as Kyn. This promotes immune tolerance in tumor cells and protects them from immune attacks 120 .
Although some viewpoints suggest that cancer cells compete with T-cells for glucose, resulting in immunosuppression, some studies have found that glucose levels are not universally limited in the TME. Cells in the TME continue to have the ability to increase glucose uptake when Gln uptake is restricted. This indicates that intrinsic cellular programs play a significant role in regulating glucose uptake by immune and cancer cells, rather than being merely a matter of nutritional competition 116 . Table 5 shows the therapeutic strategies targeting AA metabolism in the TME.
Therapeutic strategies targeting AA metabolism in the TME.

Lipid metabolism
Tumor cells take up fatty acids through various fatty acid transporters, such as CD36, SLC27, and fatty acid-binding proteins (FABPs). High CD36 expression is associated with a poor prognosis in various cancers. Moreover, its expression is upregulated under various factors, such as HER2 inhibitors, high-fat diets, hypoxia, and specific oncogenes, which promote the uptake of fatty acids by tumor cells, thereby affecting tumor growth, migration, invasion, and other processes 130 .
The rapid proliferation of tumor cells increases fatty acid synthesis to meet their increased energy demands, the formation of cell and organelle membranes, and subsequent carcinogenic signal transduction73,131. Fatty acid synthase (FASN) is the rate-limiting enzyme in the biosynthesis of new lipids. Its expression is associated with advanced cancer and metastasis 132 . FASN is highly expressed in DLBCL. When the activity of FASN is inhibited, DLBCL tumor growth significantly declines and cell apoptosis increases. Reportedly, palmitoyl transferase ZDHHC21 has a tumor suppressor function in DLBCL, and targeting the ZDHHC21/FASN axis holds a potential therapeutic strategy for DLBCL 133 . Per another study, FASN is highly expressed in CTCL, and that inhibiting FASN affects the proliferation and survival of malignant cells. Moreover, FASN expression is regulated by sterol regulatory element-binding proteins (SREBP1/2), and the combined inhibition of FASN and SREBP has the potential to treat CTCL 134 .
Acetyl-CoA carboxylase (ACC), rate-limiting enzyme in fatty acid synthesis, has two isoenzymes, ACC1 and ACC2, with ACC1 being highly expressed in various cancers. SCD is a rate-limiting enzyme in the endoplasmic reticulum that catalyzes the synthesis of monounsaturated fatty acids from saturated fatty acids (SFAs). Due to their oxygen-dependent activity, cancer cells increase their dependence on an exogenous supply of unsaturated fatty acids under hypoxic conditions 130 . SCD1 is a major monounsaturated fatty acid desaturase associated with human cancers. Inhibiting SCD1 can induce the accumulation of SFAs and endoplasmic reticulum stress, thereby leading to cancer cell apoptosis. It has thus become a therapeutic target for various types of tumors 135 . The expression of SCD1 is regulated by multiple factors. Inhibition of SCD1 can also lead to ferroptosis in cancer cells, which plays an important role in tumor development 130 .
Ferroptosis is a type of iron-dependent cell death closely related to tumor progression and metastasis. Tumor cells can evade ferroptosis by maintaining a stable expression of glutathione peroxidase-4 (GPX4), thereby enhancing their metastatic potential 136 .
Key mechanisms of cholesterol metabolism
Cholesterol is a fundamental component of most cell membranes (including the plasma membrane) and its supply is crucial for cell proliferation. Cells obtain cholesterol either through de novo synthesis or by taking up cholesterol-containing lipoproteins via low-density lipoprotein receptors (LDLRs) 137 . In the cholesterol synthesis pathway, 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) and SCAP (sterol regulatory element-binding protein-activated cleavage-activating protein) are the key rate-limiting enzymes 130 . Cellular cholesterol metabolism is mainly regulated by two key transcription factors, SREBP2 and liver X receptor (LXR). SREBP2 transcribes genes involved in cholesterol biosynthesis and uptake, thereby increasing cellular cholesterol levels. LXR transcribes genes involved in cholesterol efflux and inhibits cholesterol uptake, thereby reducing cellular cholesterol levels 138 . Furthermore, cholesterol modulates membrane fluidity and the formation of lipid rafts via the mevalonate pathway, structures that are vital for tumor cell signal transduction, protein interactions, and immune surveillance 93 .
Evidence in cancer and lymphoma progression
Dysregulated cholesterol metabolism acts as a catalyst for malignancy. HMGCR is upregulated in various cancers. Its overexpression promotes cancer cell growth and migration, while its knockdown inhibits tumor occurrence 130 . Cholesterol accumulation in the TME is necessary for triggering T-cell exhaustion 139 . High cholesterol levels can disrupt the lipid metabolic network within T-cells, thereby exerting an inhibitory effect on immunity 80 . Cholesterol impairs T-cell function by inducing expression of genes associated with T-cell exhaustion, downregulating the T-cell receptor (TCR) pathway and triggering endoplasmic reticulum stress. The increase in cholesterol efflux from macrophages via C1QB induces the upregulation of immune checkpoints (PD-1, LAG-3, and TIM-3) and T-cell exhaustion by simultaneously activating XBP1/ER stress and inhibiting the TCR signaling pathway, thereby suppressing the antitumor activity of T-cells139,140. The exhaustion of CD8+ T-cells caused by immunosuppressive TME is a major obstacle affecting the efficacy of immunotherapy. CD8 T-cell exhaustion has been reported in various hematological malignancies, including AML, ALL, CLL, MM, and lymphoma 140 . In lymphoma specifically, metabolic dysregulation correlates with progression. In DLBCL, SOX9 promotes cholesterol synthesis by upregulating the expression of DHCR24. Knockdown of SOX9 increases apoptosis, while simultaneously downregulating the expression of the anti-apoptotic proteins Bcl-xL, Mcl-1 and Bcl-2, and upregulating the expression of the pro-apoptotic proteins Bad and Bax. A high-cholesterol environment can inhibit apoptosis, and the inhibition of cholesterol synthesis can induce apoptosis 141 . For anaplastic large cell lymphoma (ALCL), a classic study has demonstrated that squalene metabolism serves as a key regulatory node in tumor progression: ALCL cells exhibit abnormally elevated squalene accumulation via FDFT1 activation, which not only scavenges lipid peroxides to enhance oxidative stress resistance but also suppresses ferroptosis through the GPX4/ACSL4 axis. In addition, squalene metabolites can potentiate STAT3 signaling to facilitate tumor cell proliferation and immune evasion 142 .
Therapeutic relevance and targets
Targeting cholesterol pathways offers promising strategies for lymphoma treatment. Per previous studies, in cell lines, such as ALL and CLL, the use of statins to inhibit cholesterol synthesis not only inhibits cell proliferation and induces apoptosis but also enhances the efficacy of chemotherapeutic drugs. Moreover, targeting cholesterol uptake receptors, such as SCARB1, or regulating the activity of cholesterol synthesis-related transcription factors (such as SREBP2 and LXRs), affect the survival and proliferation of lymphoma cells 143 . For ALCL, targeted inhibition of squalene metabolism has shown significant therapeutic potential: combined use of FDFT1 inhibitors and ferroptosis inducers can synergistically suppress ALCL growth by disrupting oxidative stress tolerance and lipid homeostasis 142 . Moreover, statins can lower cholesterol levels and disrupt the function of lipid rafts, thereby affecting the signaling pathways related to tumor cell proliferation 93 . Collectively, these findings establish a robust theoretical basis for developing targeted therapies against lymphoma by exploiting cholesterol metabolism.
Key mechanism: lymphoma cells harness lipid metabolism to survive oxidative stress
Fatty acid β-oxidation (FAO) is an important pathway for fatty acid degradation in mitochondria. It alleviates ROS levels by generating Nicotinamide Adenine Dinucleotide Phosphate (NADPH) and helps tumor cells survive in oxidative stress environments. Some tumor cells, especially invasive and metastatic cells, may have adaptive advantages in response to oxidative stress. They can upregulate antioxidant defense systems such as superoxide dismutase (SOD), GPX, and catalase to counteract the damaging effects of ROS and enable tumor cells to survive and thrive in environments characterized by oxidative stress 136 . Notably, in ALCL, squalene metabolism represents a unique oxidative stress adaptation mechanism: accumulated squalene acts as an endogenous lipid antioxidant to reduce lipid peroxidation, and genetic or pharmacological inhibition of FDFT1 can sensitize ALCL cells to ferroptosis inducers (e.g. ML162, RSL3) 142 . This finding further supports that cholesterol metabolic intermediates, beyond their structural roles, exert pro-tumorigenic effects through non-metabolic regulatory pathways. In BL, the MYC-induced transcription factor SREBP1 drives the expression of enzymes related to fatty acid synthesis and cooperates with MYC-induced glycolysis and Gln breakdown to provide acetyl-CoA for lipid synthesis. Similarly, MYC induces phosphocholine synthesis in DLBCL by transcriptionally activating phosphocholine cytidylyltransferase 1A (PCYT1A). Inhibition of lipid synthesis can impair MYC-dependent lymphoma growth 144 .
Evidence in cancer: correlation of MYC abnormalities with aggressive lymphoma phenotypes
Carcinogenic MYC aberrations such as genomic translocations are prominent features of B-cell lymphomas. Elevated MYC levels regulate fatty acid uptake and promote FAO, providing sufficient energy under conditions of limited nutrient and oxygen availability, thereby accelerating lymphoma cell proliferation. In BL, MYC gene rearrangement leads to the upregulation of fatty acid synthesis-related genes, and cells enhance FAO to meet their energy demands. Inhibition of fatty acid metabolism-related enzymes (such as CPT1) can exert cytotoxicity in cancer cells 143 . The high expression of PRMT5 promotes progression of MCL. PRMT5, a key regulatory factor that activates lipid metabolic reprogramming through MYC, participates in the growth and survival of MCL. This induces the expression of SREBP1/2 and FASN through the MYC pathway to promote the growth and expansion of MCL. Moreover, high expression of PRMT5, SREBP1/2, and FASN is closely associated with a poor prognosis in patients with MCL 145 . Beyond protein regulation, phospholipids play a crucial role in the initial signal transduction of B-cell activation and malignant transformation. Among them, PIP3 activates the PI3K/AKT signaling pathway, promoting the survival, proliferation, differentiation, and migration of B cells. The PI3K/AKT pathway is often abnormally activated in lymphomas. In DLBCL and MCL, mutations in PI3K or PTEN lead to increased PIP3 levels, thereby enhancing the kinase activity of AKT and promoting the proliferation, progression, and survival of cancer cells 143 . In addition, in patients with DLBCL, high expression of CAMKIIδ is associated with poor prognosis, and its absence inhibits lipolysis, leading to insufficient energy supply and suppression of tumor growth 146 .
Challenges and unresolved issues in metabolic adaptation
While the interplay between MYC, lipid metabolism, and oxidative stress resistance is increasingly clear, further inquiry is needed to fully elucidate how varying microenvironmental constraints influence these pathways across different lymphoma subtypes. Understanding the precise thresholds at which metabolic inhibition becomes lethal to tumor cells, versus when it might trigger compensatory survival mechanisms, remains an open question crucial for optimizing therapeutic strategies. Table 6 shows the therapeutic strategies targeting lipid metabolism in the TME.
Therapeutic strategies targeting lipid metabolism in the TME.

Discussion
Linking TME metabolism and cell therapy – mechanisms, challenges, and future directions
This review systematically elaborates on the reprogramming of glucose, AA, and lipid metabolism within the TME and its profound impact on the function of immune components (e.g. T-cells, macrophages, MDSCs). These metabolic features are not only drivers of tumor progression but also critical determinants of the success or failure of novel cell-based therapies, including chimeric antigen receptor T-cell (CAR-T) therapy. Notably, the TME is classified into hot and cold phenotypes based on immune characteristics, intersecting with metabolic reprogramming to shape therapeutic responses. Hot tumors have a TME rich in tumor-infiltrating lymphocytes (TILs), PD-L1 overexpression, genomic instability, and preexisting antitumor immunity, making them favorable for and responsive to ICIs alone. In contrast, cold tumors exhibit a low inflammatory signature, absent intratumoral CD8+ T-cells, a low immunoscore, and no effective endogenous antitumor response, rendering ICIs alone ineffective. Treating cold tumors requires combination therapies to recruit immune cells and convert them into hot tumors for immunotherapy to work 147 . Integrating TME metabolic mechanisms with cell therapy strategies, on the basis of distinguishing hot and cold TME phenotypes, represents a core approach to overcoming current therapeutic bottlenecks and developing next-generation treatments.
Metabolic reprogramming in the TME creates a nutrient-scarce, metabolically hostile niche that directly impairs the function and persistence of adoptively transferred cells (e.g. CAR-T cells, NK cells) 115 . Tumor cells and stromal components compete fiercely for glucose, Gln, and arginine, leading to nutrient depletion and metabolic exhaustion of therapeutic cells73,120. For instance, the Warburg effect-driven lactic acid accumulation in the TME not only acidifies the extracellular environment to inhibit T-cell proliferation and cytotoxicity but also polarizes TAMs toward the M2 phenotype, forming a feedforward loop of immunosuppression 73 . Conversely, metabolic reprogramming of TME components also presents actionable targets to enhance cell therapy. For example, inhibiting glycolytic enzymes (HK2, PFKP) or glutaminase (GLS) in tumor cells can alleviate metabolic competition, improving the metabolic fitness of therapeutic cells97,108,127. Targeting cholesterol metabolism—such as suppressing squalene accumulation in ALCL or using statins to disrupt lipid raft formation—can reverse T-cell exhaustion and enhance CAR-T cell infiltration138,142.
The immune landscape of the TME is a key determinant of cell therapy success. TAMs, MDSCs, and CAFs collectively create an immunosuppressive barrier that attenuates the activity of therapeutic cells. M2-polarized TAMs express PD-L1, IDO, and IL-4I1, which directly inhibit T-cell function or induce T-cell exhaustion through ligand-receptor interactions (e.g. PD-L1/PD-1) and metabolic remodeling (e.g. Trp catabolism)7,73. MDSCs further reinforce immunosuppression by secreting ARG1 and iNOS41,120. These interactions contribute significantly to cell therapy resistance. In R/R DLBCL, MDSCs induce M2 macrophage polarization and inhibit NK cell maturation, reducing the efficacy of CAR-T therapy48,49. However, targeting these immune components can sensitize lymphoid malignancies to cell therapy. For example, combining CAR-T cells with CSF1R inhibitors (to deplete M2 TAMs) or JAK inhibitors (to reduce MDSC abundance) has shown synergistic antitumor effects in preclinical models11,53. Similarly, anti-CD47 antibodies enhance the phagocytic activity of TAMs, promoting the clearance of lymphoma cells and improving the persistence of therapeutic cells5,14.
Beyond conventional CAR-T, emerging cell therapies are actively exploring the proactive utilization of metabolic pathways. M2-type TAMs are a major immunosuppressive and metabolically competitive component in the TME. Engineering CAR-M (macrophages) to skew toward a pro-inflammatory M1 phenotype, or concurrently knocking out immunosuppressive metabolic enzymes (e.g. ARG1), holds promise for transforming them from “accomplices” into “allies,” directly reshaping the metabolic and immune landscape of the TME. Future CAR designs may incorporate sensor modules responsive to specific TME metabolites (e.g. high potassium, low glucose, high lactate). This would allow for precise regulation or enhancement of CAR-T cell activation or function within the immunosuppressive metabolic milieu, enabling a more intelligent and adaptive response.
Despite the promising outlook, this field faces multiple challenges, which are further complicated by the heterogeneity of hot and cold TME phenotypes: metabolic dependencies may vary significantly across different lymphoma subtypes and even within the same tumor, and the metabolic characteristics of hot and cold TMEs add an additional layer of complexity to this heterogeneity. Tumor cells possess metabolic plasticity, potentially rapidly activating alternative pathways when one pathway is blocked, leading to the failure of single-target metabolic strategies. The metabolic state of the TME is dynamic, changing with disease progression, post-treatment, and across different anatomical sites. Determining the optimal timing and sequence for combining metabolic interventions with cell therapy requires further in-depth exploration. Metabolic modulators, while inhibiting tumor and immunosuppressive cells, may also have unforeseen negative effects on the CAR-T cells themselves.
Conclusion
The TME plays an extremely crucial role in the occurrence, development, drug resistance, and immune escape of lymphomas. Immune cells mediate immune suppression, the non-immune microenvironment participates in tumor progression, and abnormal metabolic activities in tumor cells confers growth advantages. In summary, the lymphoma TME is an ecosystem of aberrantly active metabolism, where metabolic reprogramming is a core mechanism driving immunosuppression and therapy resistance. A deep understanding of these metabolic interactions provides novel perspectives and targets for optimizing existing CAR-T therapies and designing next-generation cell therapy products.
Footnotes
Acknowledgements
Not applicable.
Ethical considerations
Not applicable.
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by Tianjin Natural Science Foundation Project (grant no. 25JCZDJC00830).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Not applicable.
Statement of human and animal rights
This article does not contain any studies with human or animal subjects.
Statement of informed consent
There are no human subjects in this article and informed consent is not applicable.