
Abstract
Objective
To use the results of the first five years of a cystic fibrosis newborn screening program to estimate the cystic fibrosis birth prevalence and spectrum of cystic fibrosis transmembrane conductance regulator (CFTR) gene variants in Yucatan, Mexico.
Methods
Screening was performed from 2010 to 2015, using two-tier immunoreactive trypsinogen testing, followed by a sweat test. When sweat test values were >30 mmol/L, the CFTR gene was analyzed.
Results
Of 96,071 newborns screened, a second sample was requested in 119 cases. A sweat test was performed in 30 newborns, and 9 possible cases were detected (seven confirmed cystic fibrosis and two inconclusive). The most frequently detected CFTR pathogenic variant (5/14 cystic fibrosis alleles, 35.7%) was p.(Phe508del); novel p.(Ala559Pro) and p.(Thr1299Hisfs*29) pathogenic variants were found.
Conclusions
Cystic fibrosis birth prevalence in southeastern Mexico is 1:13,724 newborns. Immunoreactive trypsinogen blood concentration is influenced by gestational age and by the time of sampling. The spectrum of CFTR gene variants in Yucatan is heterogeneous.
Introduction
Cystic fibrosis (CF) has an estimated newborn prevalence of 1:4000. 1 It is infrequent among Indian (1:40,000–100,000) and Japanese (1:350,000–1:1,000,000) newborn populations, 2 but in the Hispanic population of California, birth prevalence is 1:9259. 3 In Latin America, only Costa Rica, Paraguay, and Uruguay have implemented nationwide mandatory newborn screening (NBS) for this disease, and in Mexico, screening is available only through some regional NBS programs or privately, 4 hence CF birth prevalence in this region is poorly characterized. In developed countries, CF patients currently have a life expectancy of ∼40 years, 5 an outcome achieved through combining better treatments and NBS strategies that allow earlier medical management.6-8 However, even in most developed countries, where neonatal detection of CF has been recommended and adopted, testing procedures are complex, and analytical protocols are not uniform. 1
Immunoreactive trypsinogen (IRT) has been used as a CF biomarker since Crossley et al. first demonstrated that trypsinogen levels are higher in newborns with CF. 9 Interpretation of IRT tests is complex, due to confounding factors such as age-related declines in IRT blood levels, prematurity, and perinatal stress.1,10 All newborn CF screening strategies begin with IRT quantification as the first tier, followed by a second IRT determination or quantitation of pancreatitis-associated protein and/or CF transmembrane conductance regulator gene (CFTR, MIM*602421) analysis as a second tier, usually with a specific panel of the more prevalent pathogenic variants from the screened population. A complete CFTR gene study by automated Sanger or next-generation sequencing, with or without multiplex ligation-dependent probe amplification analysis, has also been used as a final tier.3,5,6,9–12 Other metabolites, such as fecal elastase, have been proposed as alternative biomarkers. 13 Independent of the screening protocol, all infants with a positive screening test require further study. 14 Current guidelines state that a CF diagnosis can be confirmed in individuals identified by NBS with sweat chloride test (ST) values ≥ 60 mmol/L at any age or in the range of 30–59 mmol/L, if two pathogenic variants of the CFTR gene are documented.15,16 However, in some cases, other functional studies, such as nasal potential difference and intestinal current measurements, are needed. 17
Molecular CFTR gene analyses are routinely performed in almost all NBS laboratories in developed countries, and most use specific mutational panels developed for the genetic population background.3,11,18,19 These tests are expensive and sometimes unaffordable, and in low- and middle-income countries, access to them is limited. Over 2000 different pathogenic variants of the CFTR gene have been reported, with distinct prevalence among diverse ethnic groups. 20 Mexican symptomatic CF patients have one of the widest CFTR mutational spectra worldwide, with the “delta-F508” variant (formerly NM_000492.3:c.1521_1523del or p.(Phe508del) rs113993960), the most common pathogenic variant, accounting for 44% of CF alleles. 21 However, because of the ethnic diversity and heterogeneity of genetic backgrounds in the Mexican population, 22 frequencies of CFTR mutations may vary according to geographical region. In northeastern Mexico, the allelic frequency of the p.(Phe508del) variant is 61.5%, 23 which is higher than that described in any other Latin American population (e.g., 22.9% in Costa Rica, 59.1% in Argentina 24 ). In Yucatan, an area with a high Mayan ancestry, the frequency of CFTR alleles remains unknown.
This study aimed to present the results of the first five years of a Mexican CF screening program, showing for first time the birth prevalence and the spectrum of CFTR gene variants in Yucatan, Mexico.
Methods
A mandatory expanded NBS protocol testing for congenital hypothyroidism, aminoacidopathies, organic acidemias, fatty acid oxidation defects, congenital adrenal hyperplasia, biotinidase deficiency, galactosemia, hemoglobinopathies, and CF was applied to all newborns delivered at Health Facilities for Infant-Maternal Care at the Health State Ministry of Yucatan in southeastern Mexico, between 2010 and 2015. Five drops of blood were taken from the heel and collected on standardized filter paper (Guthrie cards). Demographic data were obtained, and all confirmed cases received professional genetic counseling.
The IRT/IRT/ST screening strategy is shown in Figure 1. For IRT blood values > 70 ng/mL and < 250 ng/mL, a second blood sample was requested. An ST was requested for all newborns with a second cutoff IRT blood concentration > 60 ng/mL. Cases with an original IRT of > 250 ng/mL were sent directly to ST.
Cystic fibrosis newborn screening algorithm and overview of results. CF: cystic fibrosis; NBs: newborns; IRT: immunoreactive trypsinogen.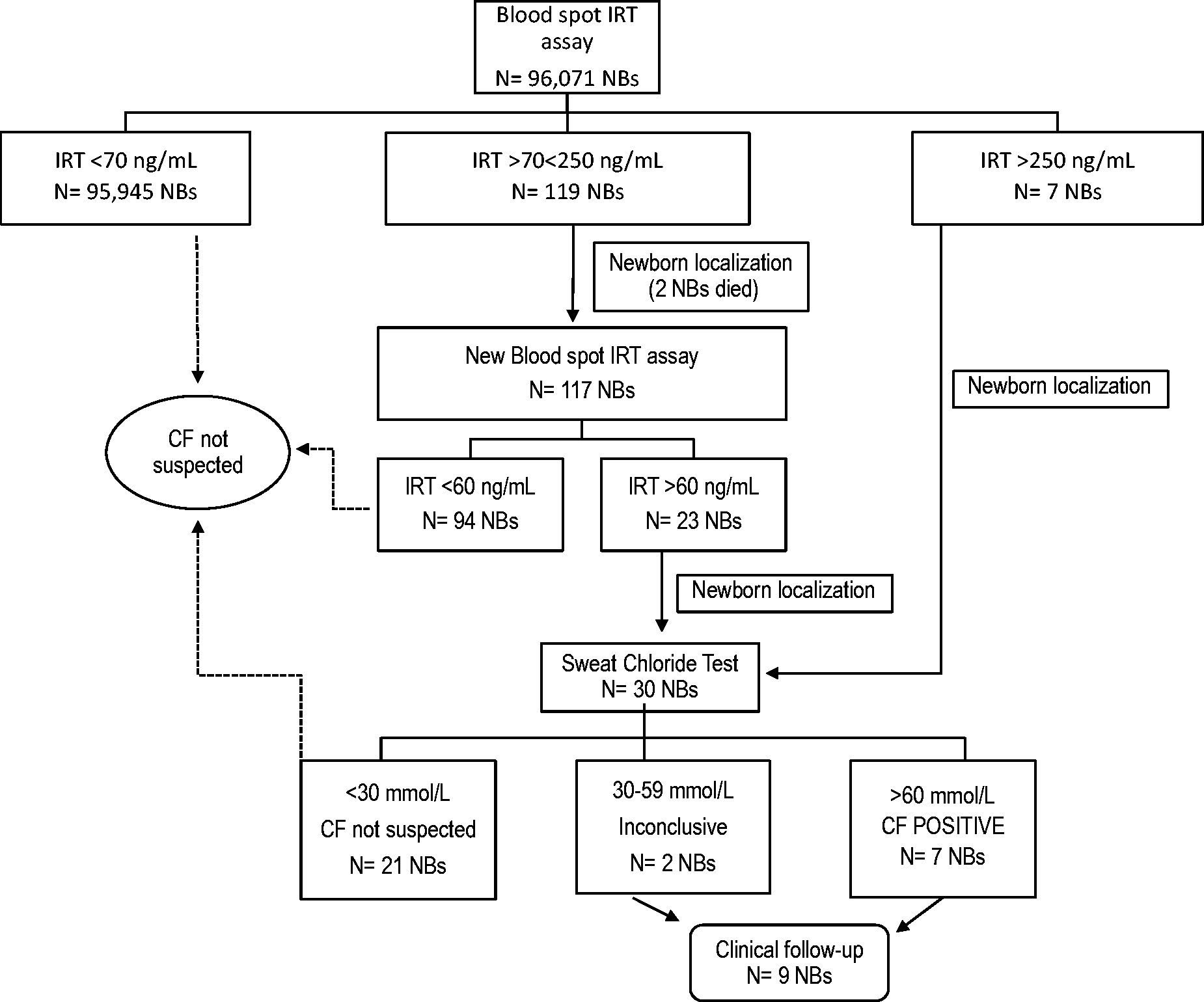
IRT was measured in blood spot samples by time-resolved immunofluorometric assay using an Autodelfia instrument and commercial kits (Perkin Elmer, Waltham, MA). Sweat samples were obtained using the Macroduct® (Webster Sweat Collection System 3700-SYS, Wescor). After cleansing the skin with water, pilocarpine iontophoretic was used to stimulate sweat production (pilogel pads). Sweat chloride concentration was measured in the laboratory according to the CLSI guidelines. 10 Newborns with sweat chloride concentrations > 60 mmol/L were considered true CF cases, and those with values between 30 and 59 mmol/L were considered inconclusive cases, according to 2015 CF Foundation diagnosis consensus conference recommendations on the diagnosis of CF in screened populations. 15 All true or inconclusive CF cases were subsequently referred to the Dr Agustín O’Horán Hospital for medical evaluation and follow-up.
DNA extracted from dried blood spots from nine newborns considered true or inconclusive CF cases was subjected to CFTR analysis after obtaining written informed parental consent. In eight cases, Sanger bidirectional automated sequencing of the 27 coding exons of the CFTR gene (NG_016465.4, RefSeqGene, LRG_663, NM_000492.3) and two intronic fragments (to identify the pathogenic variants “1811 + 1.6kbA>G” [rs397508266] and “3849+10kbC>T” [rs75039782]) was performed (DNA-GEN, S.C. Laboratory). The remaining patient was analyzed against a panel of 32 pathogenic variants (Test Code 10458; Quest Diagnostics Nichols Institute, San Juan Capistrano, CA). 25 Previously unreported missense CFTR variants identified by sequencing were subjected to in silico analysis with the PolyPhen 26 , SIFT, 27 and MutationTaster 28 programs to predict their damaging effects.
Results
A total of 96,071 newborns in Yucatan (45.6% females and 54.4% males), representing 97% coverage of annual births, were screened for IRT over the five-year period. The modal age at sampling was five days (range, 1–28 days); 76.6% of samples were taken during the first week of life, 18.4% in the second, and 3.6% and 1.4% in the third and fourth weeks, respectively. A total of 7.7% of newborns were premature (<36 weeks gestation).
Figure 2(a) and (b) shows the IRT values in blood samples obtained in the first week of life from full-term and preterm newborns. The median IRT blood concentrations in first-week samples were 15.30 ng/mL in full-term newborns and 14.6 ng/mL in preterm newborns. A significant difference using Mann-Whitney U test between the IRT values of the full-term versus preterm newborns was found only for those samples obtained in the first and second weeks of life (Figure 2(c)).
Distribution of the neonatal IRT values in the first week of life by newborn gestational age (a) Full term, (b) Pre-term, and (c) IRT concentration values by gestational age according to the different weeks of age at the time of blood sampling. IRT: immunoreactive trypsinogen; T: full-term; P: pre-term.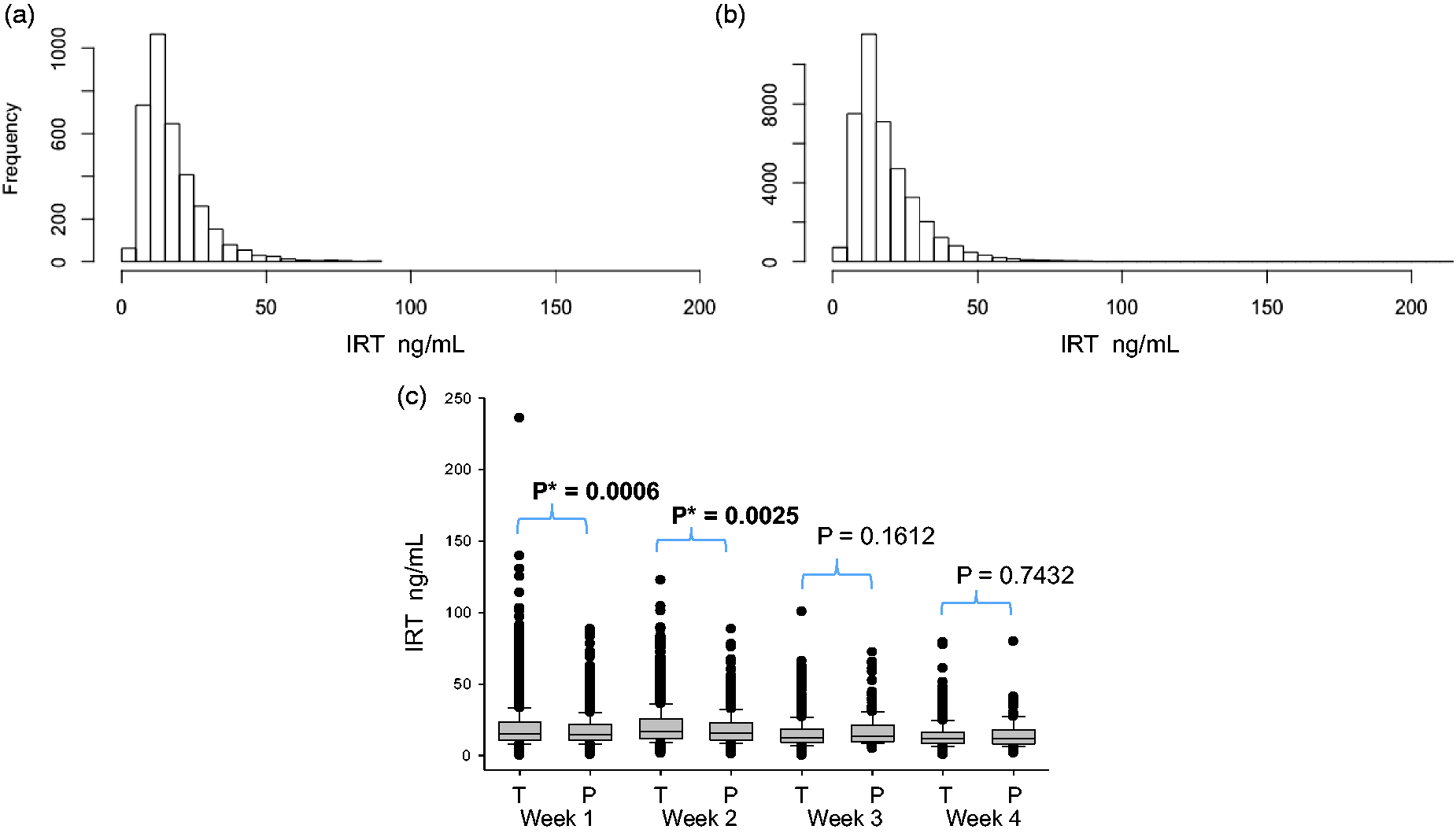
An overview of the screening strategy and results are shown in Figure 1. The cutoff value of 70 ng/mL represents the cumulative 99.5th percentile of the first 2500 studied newborns. The cutoff value for the second sample was determined based on the age of the newborn at sampling. Among the 96,071 screened newborns, blood samples from 126 were above the cutoff level (70 ng/mL), corresponding to a recall rate of 0.12%. In two newborns with high IRT blood concentrations (147 and 225 ng/L), a second sample was not obtained because the newborns died from unknown causes soon after the first sample was taken. In this study, seven CF cases were confirmed by the IRT/IRT/ST algorithm strategy, yielding a birth prevalence of 7.2 per 100,000 newborns (1:13,724 newborns).
IRT blood concentration, sweat chloride values, sweat test interpretation, NBS CF diagnosis, and spectrum of CTRF gene variants in the studied southeastern Mexican population.

CF: cystic fibrosis; IRT: immunoreactive trypsinogen; NBS: newborn screening.
Complete sequencing of the CFTR gene 29 was performed in all patients, except patient 5, for whom a 32-mutation panel 25 was used. Variant nomenclature at complementary DNA; cDNA and protein levels is according to the reference sequences NM_000531.5 and NP_000522.3, respectively. New variants are shown in bold type.
Discussion
We used a simple IRT/IRT/ST strategy (Figure 1) to detect seven CF cases. In this work, we had an extremely low recall rate (0.24%–0.9%) compared with that reported elsewhere.1,31,32 The explanation for this may be related to the ethnic characteristics of the population, with the small screened sample (less than 100,000 newborns), or with our cutoff value (70 ng/mL), which is higher than that used in other programs (50–62 ng/mL).1,3
Prior to our study, the occurrence of CF in Mexico had been estimated mainly based on the number of subjects diagnosed with symptoms (∼1:8,500) 2 , but details of the geographic origins of cases were not specified. In a large multiethnic NBS study in California, Kharrazi et al. 3 reported differing CF birth prevalences, with the lowest frequency in the Hispanic population (1:9,259), but the term “Hispanic” included not only the Mexican population but also individuals from Central and South America with different genetic backgrounds. Our work is the first determination of birth prevalence of CF using population-based NBS data, finding 7.2 affected per 100,000 newborns (1: 13,724 newborns) in Yucatan. The Mexican contemporary population represents a complex admixture of European, Native American, and African, with important ethnic variations throughout the country. 22 Accordingly, CF birth prevalence and CFTR variants would be expected to differ among geographic regions.
No false-negative results have been found to date, but not all late-diagnosed CF cases are relayed to the screening laboratory because of the absence of a national surveillance system for this disease.
The ideal time for blood sampling for CF NBS is in the first days of life, 1 but in some circumstances, newborns can only be screened later. In our study, 76.6% of newborns were screened during the first week of life. In Mexico, the official recommendation for NBS sampling is between age three and five days. Our results show that four of seven positive CF cases were sampled at those ages. In the remaining three newborns, the blood sample was obtained later (age six to seven days). In this study, the average age at CF diagnosis confirmation was 1.3 months (Table 1), which is acceptable according to the Laboratory Guide to NBS for CF in the UK and is similar to that in other programs.19,33
As previously described,1,14 we found that IRT blood concentrations were age dependent, with lower values at older ages (Figure 2(c)). Prematurity, low birth weight, and perinatal stress have been also identified as factors that affect IRT blood levels. 1
In our study, two ill newborns had an initially high IRT blood concentration and subsequently died, preventing further studies. In these two cases, the diagnosis of CF seems unlikely, because elevated IRT blood values have been observed among CF-unaffected newborns admitted to neonatal intensive care units, 1 although a diagnosis of CF cannot be discarded.
CFTR allelic heterogeneity has been described in late-diagnosed CF patients in Mexico, 21 but our study provides information about the mutational spectrum of CF patients identified through a NBS program applied to a southeastern Mexican population. The mutation detection rate in the seven CF cases identified by the IRT/IRT/ST strategy was 85.7% (12/14 CF alleles; Table 1), and the most common pathogenic variant was p.(Phe508del) (5/14 CF alleles, 35.7%). Our results are similar to the findings of Kharrazi et al., 3 where this variant was the most frequent (45%) in the Hispanic population.
The only previous data about the CFTR variant distribution in regions of Mexico come from patients from northeastern Mexico, where the frequency of the p.(Phe508del) variant (61.5%) is closer to that observed in Europe (70%). 20 The characterization of pathogenic variants and CFTR genotypes among CF individuals belonging to specific ethnic groups is important for designing the best mutational panel to use as a second tier molecular diagnostic test, establishing treatments and predicting prognosis of patients.3,34
Given the diversity of known and novel CFTR mutations, molecular diagnosis using commercially available panels could be of limited value if these panels are not designed for a specific population. In this study, only two pathogenic variants, p.(Phe508del) and p.(Gly542Ter), which represented 50% (7/14) of the total CF alleles identified by complete sequencing of the CFTR gene, would have been detected using the 32-mutation commercial panel. 25 Genotyping more CF patients from this southeastern Mexican population by direct sequencing would be necessary to design a population-specific panel of CFTR mutations containing pathogenic variants that are not included in most common panels based on the mutational spectrum identified in other populations (e.g., Caucasians). Our data suggest that it should be possible to identify ∼85% (12/14) of CF alleles from the southeastern Mexican population by analyzing the seven pathogenic variants characterized herein, but the mutational spectrum could change, as the study sample increases.
In this study, two novel CFTR variants were found. The c.1675G>C or p.(Ala559Pro) variant identified in a patient with consanguineous parents affects a position within the ATP-binding cassette domain of the CFTR protein (amino acids 389 to 670) that is highly phylogenetic conserved from yeast to humans. At least 599 CF variants, including other CF-causing variants at the same p.Ala559 position [i.e., p.(Ala559Thr), p.(Ala559Glu) and p.(Ala559Val)] 35 affect the nucleotide-binding domain, which carries out the ATP-binding and hydrolysis required for opening and closing of the gate. Not surprisingly, most CF-causing mutations occur in this domain. Accordingly, the novel p.(Ala559Pro) variant probably corresponds to a class III mutation that produces a protein with no residual CFTR function and a severe phenotype. 5 The other new frameshift microdeletion, c.3895del, encoding p.(Thr1299Hisfs*29), could be considered a class I severity variant with absent or reduced synthesis of CFTR protein.
The p.(Lys536Glu) [rs148173473] variant was identified in one CF case (Table 1, patient 7) with abnormal and categorically positive ST (>70 mmol/L), but no other pathogenic CFTR variants were identified after complete sequencing of the CFTR gene. Although this change lies in the ATP-binding cassette domain of the CFTR protein, to our knowledge, there is no published evidence to support its pathogenicity, and an in silico analysis of this variant was inconsistent because p.Lys536 is not highly conserved, even among mammals. The Exome Aggregation Consortium has described the p.(Lys536Glu) variant only in a heterozygous state in the Latin American population at very low allelic frequency (0.000792). 36 For these reasons, we consider the p.(Lys536Glu) variant to have uncertain clinical significance. Further characterization of the responsible genotype could rely on multiplex ligation-dependent probe amplification or next-generation sequencing-based strategies, as gross gene deletions/duplications accounted for 17% of unidentified CF alleles by Sanger sequencing in non-white individuals. 29
Molecular analyses also allowed us to detect one CF carrier (Table 1, patient 8), who subsequently received genetic counseling. Detecting carriers is a controversial issue, with some authors considering this situation to be an unwanted “side effect” of molecular testing of CF through NBS. 6
Conclusion
Using an IRT/IRT/ST strategy, we detected seven newborn CF patients, yielding a birth prevalence of 1:13,724 in southeastern Mexico. As with previous reports, IRT blood concentration was influenced by gestational age and by time of sampling. In the studied population, a CFTR molecular study identified 85.71% (12/14) of CF alleles, which were distributed among seven different variants, including two novel CFTR pathogenic alleles p.(Ala559Pro) and p.(Thr1299Hisfs*29). The p.(Phe508del) mutation was the most frequent documented CF allele (35.7%, 5/14).
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: VAM and IGI have received honorarium as TamizMas® de Químicos Maldonado technical advisors. GAA and AOMA have received payment for the CFTR gene molecular analyses performed at DNA-GEN, S.C., through TamizMas® de Químicos Maldonado. FJCG, LAHP, PMC, CMMG, and FAMS disclose employment by TamizMas® de Químicos Maldonado. The other authors have no interests to declare.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was by sponsored by TamizMas® and by the Sociedad Mexicana de Errores Innatos del Metabolismo y Tamiz Neonatal, A.C.